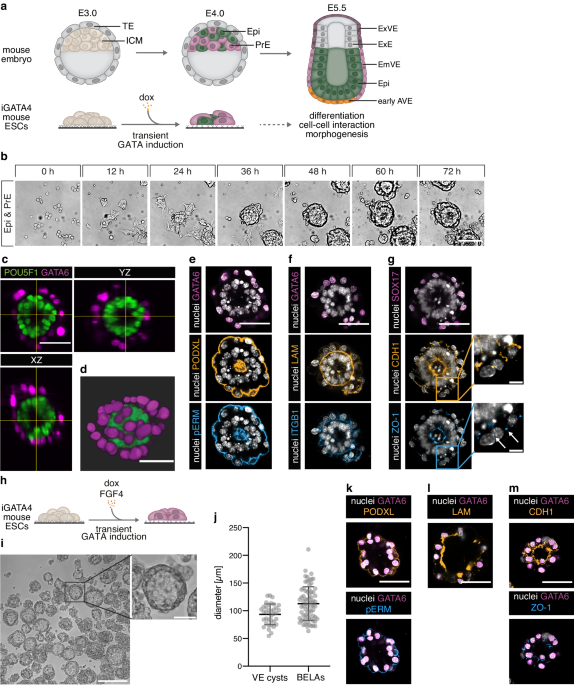
Cell lines
All embryonic stem cell lines used in this study were on an E14tg2a background31. The inducible Tet::GATA4-mCherry (iGATA) lines have previously been described9. We used two different clones in this study, C5 and C6, that differ in their induction rate. ESCs were maintained on fibronectin-coated dishes in an N2B27-based medium supplemented with 1 µM PD0325901 (SeleckChem), 10 ng/ml LIF (protein expression facility, MPI Dortmund), and 3 µM CHIR99021 (Tocris), referred to as 2i + LIF32. N2B27 was prepared as a 1:1 mixture of DMEM/F12 and Neuropan Basal Medium (both from PAN Biotech), supplemented with 1X N2 and 1X B27 supplements, 1X L-Glutamax, 0.0025% BSA, and 0.2 mM ß-mercaptoethanol (all from ThermoFisher). All iGATA4 cell lines were kept under constant selection with 200 µg/ml G418 (Sigma) to prevent silencing of the inducible transgene. The XEN cell lines IM8A1-GFP33 and X1034 have previously been described and were kindly shared by Kat Hadjantonakis. XEN cells were maintained in a GMEM-based medium, supplemented with 10% fetal bovine serum (FBS), 2 mM GlutaMAX, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol and 10 ng/mL LIF. All cell lines were cultured at 37 °C with 5% CO2, and regularly tested for mycoplasma contamination.
Mouse strains
Animal experiments and husbandry were performed according to the German Animal Welfare guidelines and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (State Agency for Nature, Environment and Consumer Protection of North Rhine-Westphalia). The mice used in this study were at age from 6 weeks to 5 months. The animals were maintained under a 14-hour light/10-hour dark cycle with free access to food and water. Male mice were kept individually, whereas the female mice were housed in groups of up to four per cage.
Mice used for tetraploid complementation were of the B6C3F1 or CD1 strains and were raised in-house. Mice carrying the TCF/Lef:H2B-GFP reporter allele have previously been described19. To obtain TCF/Lef:H2B-GFP embryos, heterozygous TCF/Lef:H2B-GFP stud males were crossed with CD1 females.
Generation of mutant and transgenic ESC lines
To generate a Cer1 reporter in the iGATA cell line, the Cer1 promoter region 4 kb upstream of the start codon was amplified from genomic DNA. A puromycin resistance cassette and a H2B-Venus sequence were amplified from Sprouty4 targeting vectors described in Morgani et al.35, and Raina et al.9. All three fragments were cloned via Gibson assembly using a HiFi DNA assembly kit (NEB) into a vector backbone containing PiggyBac transposition sites36. The Cer1:H2B-Venus reporter construct was co-transfected with CAG-pBASE36 using Lipofectamine 2000 (ThermoFisher) according to manufacturer’s instructions. Cells were selected with 1.5 µg/ml puromycin (Sigma) starting 24 h after transfection. Colonies were picked one week after transfection, expanded, and evaluated for co-localization of Cer1 reporter activity and Cer1 mRNA.
CRISPR/Cas9 was used to mutate the Nodal locus in iGATA ESCs (clone C6) and one subclone carrying the Cer1:H2B-Venus reporter construct. sgRNAs 5’-CCCCATGGACATACCCACTG-3’ and 5’-CCAGTCGAGCAGAAAAGTGT-3’ defining a 244 bp region in Nodal exon 2 were cloned into pX458 (Addgene plasmid #48138) or pX459 (Addgene plasmid #48139) using BbsI (NEB) according to Ran et al.37. Cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. To enrich for transfectants, cells were either selected with 1.5 µg/ml puromycin for 2 days, or flow sorted for GFP-expression before seeding at clonal density. We established several clonal lines, and used primers 5’-GTGGACGTGACCGGACAGAACTG-3’ and 5’-GGCATGGTTGGTAGGATGAAACTCC-3’ to PCR-amplify a sequence around the CRISPR mutation site. Clones that gave a shortened amplicon compared to the wild type were chosen for further analysis, and the exact sequence of the mutated alleles was determined by Sanger sequencing.
To generate constitutively labeled cell lines, we modified a piggybac vector for the constitutive expression of H2B-Cerulean38 by either replacing its puromycin resistance cassette with a blasticidin resistance cassette from pCX-H2B-Cerulean-IRES-bsd39 using restriction enzymes PmiI and PstI, or by replacing the H2B-Cerulean sequence with an mCherry coding sequence using restriction enzymes SpeI and NotI. Vectors were co-transfected with CAG-pBASE36 using Lipofectamine 2000 according to the manufacturer’s instructions, and transfected cells were selected with 15 µg/ml blasticidin 48 h after transfection. Four days after transfection, cells were flow sorted for the expression of fluorescent proteins, and seeded a clonal density. Several clones were expanded, and two to three suitable clones with homogeneous, moderate H2B-Cerulean and/or mCherry fluorescence were selected by epifluorescence microscopy for further experiments.
Differentiation of pure cultures of PrE cells and subsequent AVE differentiation
Pure cultures of PrE cells from iGATA clone C6 were obtained by inducing with 0.5 µg/ml doxycycline in 2i + LIF for 8 h, followed by another 16 h of doxycycline treatment in N2B27 supplemented with 10 ng/ml FGF4 and 1 µg/ml heparin. To obtain these cultures from iGATA clone C5, a 4 h pulse with 0.5 µg/ml doxycycline in 2i + LIF followed by further culture in N2B27 supplemented with FGF4 and heparin was sufficient. Clone C5 was used for the data shown in Fig. 1k–m and Supplementary Fig. 2c, in all other instances, clone C6 was used.
To differentiate AVE cells from these cultures, cells were additionally treated with 50 ng/ml ActivinA upon media change from 2i + LIF to N2B27. Approximately 24 h after the start of doxycycline treatment, cells were re-seeded at a total density of 25,000 to 30,000 cells/cm2 on fibronectin-coated dishes and cultured for 3 days in N2B27 supplemented with 10 ng/ml FGF4, 1 µg/ml heparin and 50 ng/ml ActivinA.
Formation of EXE embryoids, BELAs, VE- and Epi-cysts
EXE embryoids were generated by seeding ESCs and XEN cells at a ratio of 30:70 and a final density of 30,000 cells/cm2 on dishes that had been coated with 0.1% gelatin in PBS for 30 min, followed by culture in N2B27 for 3 or 4 days.
BELAs were generated by inducing iGATA ESCs with 0.5 µg/ml doxycycline in 2i + LIF for 8 h, followed by a media change to N2B27 for 16 h. Cells were then seeded at a density of 30,000 cells/cm2 on dishes that had been coated with 0.1% gelatin in PBS for 30 min. Floating aggregates were collected for further analysis at indicated time points.
VE cysts were generated from pure cultures of PrE cells differentiated as described above, followed by re-seeding onto gelatin-coated dishes at a density of 30,000 cells/cm2 in N2B27 medium supplemented with 10 ng/ml FGF4 and 1 µg/ml heparin.
Cysts of Epi cells were made according to Bedzhov and Zernicka-Goetz, 201422, with minor modifications. iGATA ESCs were detached, resuspended in growth factor-reduced matrigel (Corning) and plated as 25 µl drops on µ-slides (ibidi). The slides were incubated at 37 °C to allow the matrigel to solidify and then filled with prewarmed N2B27 or 2i + LIF medium.
Generation of epiblast-specific Nodal knock-out embryos
Epiblast-specific Nodal knock-out embryos were generated via tetraploid complementation. Donor embryos used for tetraploid complementation were derived from the B6C3F1 strain and foster mothers for embryo transfer experiments were from the CD1 background. Briefly, tetraploid morulae were aggregated with Nodal-mutant ESCs4,38 or wild-type E14 ESCs. The aggregated embryos were cultured in KSOM (Millipore) for an additional 3 days, which were then transferred into the uterus of foster mothers. Post-implantation embryonic day (E) 5.5 tetraploid embryos were recovered by manually dissecting the uterus.
Embryo culture
The E5.5 embryos were cultured in a 4-well plate (176740, Thermo Fisher) for 24 h, in IVC (50% IVC1 and 50% IVC2) medium, at 37 °C, 5% CO2 atmosphere in air. Before use, the IVC1 and IVC2 medium was equilibrated for 30 min at 37 °C, 5% CO2 atmosphere in air. In experiments involving inhibitors treatments, the IVC medium was supplemented with pharmacological compounds: 3 μM CHIR99021 (Cat# 4423, Tocris) or 20 μM XAV (Cell Guidance Systems, Cat# SM38-10).
The composition of the IVC1 medium was previously described22,40, consisting of DMEM F-12 (21331-046, Invitrogen), supplemented with 20% heat-inactivated FCS (10828028, Invitrogen), 0.5x Pen (25 U/ml) / Strep (25 μg/ml) (P4333, Sigma), 2 nM l-glutamine (G7513, Sigma), 1x ITS-X (51500-056, Invitrogen), 8 nM β-estradiol (E8875, Sigma), 200 ng/ml Progesterone (P0130, Sigma) and 25 μM N-acetyl-l-cysteine (A7250, Sigma). The IVC2 medium22,40 was slightly modified, consisting of DMEM (12800017, Thermo Fisher Scientific) supplemented with 1.0 g/l NaHCO3 (S5761, Sigma Aldrich) 5% heat-inactivated FCS (10828028, Invitrogen), 30% (vol/vol) KSR (10828028, Invitrogen), 0.5x Pen (25 U/ml)/Strep (25 μg/ml) (P4333, Sigma), 2 nM l-glutamine (G7513, Sigma), 1x ITS-X (51500-056, Invitrogen), 8 nM β-estradiol (E8875, Sigma), 200 ng/ml Progesterone (P0130, Sigma), 25 μM N-acetyl-l-cysteine (A7250, Sigma).
Immunostaining
BELAs and VE cysts in suspension were fixed with 4% paraformaldehyde at room temperature for 1 h, washed 5 times with phosphate-buffered saline (PBS) for 5 min each, and then incubated in PBS supplemented with 1% BSA and 0.1% Triton X-100 (PBT + BSA) for 3 h at room temperature, followed by incubation with primary antibodies diluted in PBT + BSA at 4 °C overnight. Primary antibodies used were anti-Oct3/4 (POU5F1, Santa Cruz Biotechnology sc-5279 1:100), anti-E-Cadherin (CDH1, Takara M108, 1:200), anti-GATA6 (R&D, RF1700, 1:200), anti-CD29 (ITGB1, BD Pharmingen 562153, 1:100), anti-LAM (Sigma L9393, 1:500 − 750), anti-OTX2 (Neuromics GT15095, 1:200), anti-pERM (Cell Signaling Technology #3141, 1:200), anti-PODXL (R&D MAB1556, 1:200), anti-SOX17 (R&D AF1924, 1:200), anti-ZO-1 (Invitrogen 61-7300, 1:100), and anti-GFP (Abcam ab13970 1:200).
To remove the primary antibody solution, the aggregates were washed five times with PBT + BSA. Aggregates were then incubated overnight at 4 °C with secondary antibodies diluted in PBT + BSA. Secondary antibodies from Invitrogen/Life Technologies were Alexa Fluor-conjugated and used at 4 µg/ml. Nuclei were stained with Hoechst 33342 dye at 1 µg/ml (Invitrogen). The secondary antibody solution was removed by 5 washes with PBS supplemented with 0.1% Triton X-100. The aggregates were resuspended in PBS and mounted onto µ-slides (ibidi).
Epi cysts in matrigel and cells grown in µ-slides (ibidi) were stained similarly, but with extended incubation and wash times for Epi cysts, and shortened times for cells grown as 2D layers. Samples were mounted in mounting solution consisting of 16% PBS, 80% glycerol, and 4% n-propyl-gallate.
Post-implantation embryos were either fixed immediately after isolation, or after culture as described above, in 4% PFA for 20 min, and washed twice in wash buffer containing 1% fetal calf serum (FCS) in PBS. The embryos were then permeabilized in 0.1 M glycine/0.3% Triton-X in PBS for 10 min, and washed twice in the wash buffer. The embryos were then incubated with primary antibodies in blocking buffer containing 2% FCS in PBS overnight at 4 °C. Primary antibodies used were anti-Cer1 (rat monoclonal, R&D systems, Cat# MAB1986, 1:200), anti-Otx2 (goat polyclonal, R&D systems, Cat# AF1979, 1:200), anti-Oct-4A (rabbit monoclonal (D6C8T), Cell signalling, Cat# 83932 S, 1:200), and anti-GFP (chicken polyclonal, Abcam ab13970, 1:200). After two washes in wash buffer, embryos were incubated with secondary antibodies and DAPI (Carl Roth, Cat# 6335.1) in blocking buffer, which were washed twice on the next day. The stained embryos were mounted in droplets of wash buffer on 35 mm µ-dish glass bottom plates (ibidi), covered with mineral oil and stored at 4 °C until imaging.
In situ HCR
For third generation in situ HCR we used probe sets, wash and hybridization buffers together with corresponding Alexa Fluor-labeled amplifiers from Molecular Instruments41. Staining was performed according to the manufacturer’s instructions. Briefly, samples were fixed for 15 min to 1 h with 4% paraformaldehyde, washed four times with PBS with 0.1% Tween 20 (PBST) and permeabilized at least overnight in 70% ethanol at −20 °C. Samples were then washed twice with PBST, and equilibrated in probe hybridization buffer for 30 min at 37 °C. Transcript-specific probes for Otx2 (NM_144841.5), Gata6 (NM_010258) and Cer1 (NM_009887.2) were designed by Molecular Instruments. Probes were used at a final concentration of 4 nM in probe hybridization buffer and incubated overnight at 37 °C. To remove the probe solution, the sample was washed four times with probe wash buffer preheated to 37 °C and once with 5x SSC with 0.1% Tween 20 (SSCT). Samples were then equilibrated in amplification buffer for 30 min at room temperature. Alexa Fluor-labeled amplifiers were used at a final concentration of 60 nM together with Hoechst 33342 dye at 1 µg/ml and incubated overnight at room temperature. The amplifier solution was removed by six washes with 5x SSCT. Stained BELAs were resuspended in PBS and mounted on an ibidi µ-slide for imaging. 2D cultures were mounted in mounting solution consisting of 16% PBS, 80% glycerol, and 4% n-propyl-gallate.
Imaging
Cells for long-term imaging (Fig. 1b and Supplementary Fig. 1b) were seeded at a density of 30,000 cells/cm2 on 6-well plates (Sarstedt) or 8-well µ-slides plates (ibidi) and allowed to attach for 1–2 h before the start of imaging. Time-lapse movies were recorded with a 20×0.5 NA air objective on an Olympus IX81 widefield microscope equipped with a stage top incubator (ibidi), LED illumination (pE4000, CoolLED) and a c9100-13 EMCCD (Hamamatsu) camera. Hardware was controlled by MicroManager software42, and tile scans were stitched in FIJI using the pairwise stitching plugin43. Live VE cysts in Fig. 1i, j were imaged on a Leica DM IRB widefield microscope using a 20×0.4 NA (Fig. 1i) or a 40×0.55 NA (Fig. 1j) phase contrast objective.
Stained BELAs, embryos, and stained cells in 2D culture were imaged on a Leica SP8 confocal microscope (Leica Microsystems) with a 63×1.4 NA oil immersion objective.
Cultures to determine the clonal composition of AVE clusters in 2D culture (Fig. 5b) were fixed, incubated with SYTO Deep Red Nucleic Acid Stain (ThermoFisher) for one hour, and imaged with a 20×0.5 NA air objective on an Olympus IX81 widefield microscope equipped with LED illumination (pE4000, CoolLED) and an iXon 888 EM-CCD camera (Andor). Hardware was controlled by MicroManager software42 and tile scans were stitched in FIJI using the pairwise stitching plugin43.
For light sheet imaging, fixed and stained aggregates were resuspended in low melting agarose and placed in 1.5 mm U-shaped capillaries (Leica). Capillaries were placed into water filled 35 mm high glass bottom µ-dishes (ibidi). Images were acquired using an HC Fluotar L 25×0.95 NA water DLS TwinFlect 2.5 mm detection objective and an HC PL Fluotar 5×0.15 NA illumination objective on a Leica TCS SP8 digital light sheet microscope. 3D animations were created using the Leica X application suite. Z-Stack images were processed and quantified using FIJI and Imaris.
Analysis of spatial patterning in BELAs
To determine if mesoderm differentiation in BELAs was spatially patterned by Cer1:H2B-Venus expressing AVE cells, we calculated polarization vectors of the Cer1:H2B-Venus and T/Bra expression domains in BELAs according to Simunovic et al.44, We first selected BELAs for imaging that contained T/Bra+ cells one day after a 24-hour Chi pulse. Mean polarization vectors were calculated in 2D for two independent Z-slices in each BELA that were 15 µm apart in Z-direction to ensure that an independent set of cells was analyzed. In each slice, we selected Epi and VE cells based on a mask that was drawn along the Laminin-ring that separates the two compartments. Next, we segmented individual nuclei with the StarDist 2D plugin in FIJI, using the versatile (fluorescent nuclei) model with default post-processing parameters45. Under- and oversegmented cells, debris and segmentation artefacts at the boundaries of the Epi- and the VE-compartment were filtered out with size and circularity filters. We measured median fluorescence intensities per nucleus in each channel, and rescaled intensities in individual nuclei by the maximum per-nucleus median intensity in the same channel. Using this information on nuclei position and fluorescence intensity, we calculated the radius of gyration as a measure for structure size separately for each compartment, as well as the mean polarization vectors of Cer1:H2B-Venus and T/Bra expression normalized by Rgyr2 according to Simunovic et al.44. To estimate background polarization vectors in the absence of patterning, we performed randomizations per Z-slice and BELA, where we shuffled measured fluorescence intensities between nuclei positions and determined the average polarization vector of 100 randomizations for each BELA and Z-slice. This analysis revealed that not all BELAs showed a polarized Cer1:H2B-Venus domain, possibly due to the Chiron treatment which is expected to inhibit AVE differentiation, and the late stage of analysis. We categorized BELAs as AVE-polarized, when the normalized polarization vector of the Cer1:H2B-Venus domain was larger than the polarization vector in 95% of the shuffled control group. Finally, we calculated the angle between the mean polarization vector of the Cer1:H2B-Venus and the T/Bra-domains for AVE-polarized and non-polarized BELAs.
Flow cytometry
Cells for flow cytometry were detached from culture vessels, fixed in 4% paraformaldehyde for 15 min, washed with PBS and then incubated in PBS + 1% BSA + 0.25% Saponin (PBSap) for 30 min at room temperature. Afterwards, cells were incubated with primary antibodies diluted in PBSap at 4 °C overnight. The next day, cells were washed three times in PBSap and incubated with secondary antibodies diluted in PBSap for at least one hour. Cells were washed three times in PBSap, and passed through a cell strainer and analyzed immediately on a LSRII flow cytometer (BD Biosciences). Live cells were sorted on a FACS Aria Fusion (BD Biosciences). Flow cytometry data was analyzed with FlowJo (BD Biosciences).
ScRNAseq sample preparation
BELAs and VE cysts were generated as described above. Between 100 and 200 BELAs and VE cysts were manually picked under a dissection microscope for further processing. We selected round aggregates and cysts, and excluded structures that contained a large number of dead cells, or that were unusually big or small. For the VE cysts, we also aimed at excluding structures that contained a clearly visible core of putative Epi-like cells, which likely arise from insufficiently induced cells. Both BELAs and VE cysts were gently spun down, resuspended in 1 ml Accutase and incubated at 37 °C for 10 min, followed by mechanical dissociation by pipetting and further incubation in Accutase for 5 min. Next, cells were spun down, washed in PBS, and resuspended in a small volume of PBS + 0.5% BSA.
To generate Epi cysts for RNA sequencing, single cells were seeded in matrigel and cultured in 2i + LIF for one day. Then, medium was changed to N2B27, and cells were cultured for another 3 days. Cysts were recovered from matrigel by incubation in recovery solution (Corning) for 20 min on ice. Next, cysts were gently spun down and dissociated with Accutase as described for BELAs and VE cysts above. To remove residual matrigel, dissociated cells were washed once with recovery solution and twice with ice-cold PBS, followed by resuspending in a small volume of PBS + 0.5% BSA.
ScRNAseq library preparation and sequencing
Cells from all three samples were counted, and each sample was mixed with H2O and RT master mix from the Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 (10x Genomics) to obtain a cell density required for targeting 1000 (Epi and VE cysts) or 2000 (BELAs) cells. Cell suspensions were loaded on a Chromium Controller (10x Genomics) to partition cells with gel beads in emulsion. Reverse transcription, cDNA recovery and amplification, and sequencing library construction were performed according to manufacturer’s instructions (10x Genomics ChromiumNextGEMSingleCell_v3.1_Rev_D). We chose 12 PCR cycles for cDNA amplification, and 13 PCR cycles for index PCR. Concentration and insert size of sequencing libraries were determined with a BioAnalyzer High Sensitivity DNA Assay (Agilent). Libraries were sequenced by paired-end Illumina sequencing on a NovaSeq6000 instrument with a read length of 150 bp. We first performed sequencing at shallow depth with a target of 10.000 reads per cell, to confirm capturing of an appropriate number of high-quality single-cell transcriptomes. Subsequently, deeper sequencing was performed, to obtain between 100,000 and 150,000 reads per cell.
ScRNAseq data analysis
Demultiplexing, alignment to the mouse genome mm10 (GENCODE vM23/Ensembl 98, from 10x Genomics) and read quantification was performed with CellRanger (10x Genomics, v4.0.0). Subsequent analysis was carried out in R using Seurat v4.1.146. We first filtered out cells with less than 4000 different features detected and with more than 10% of the reads mapped to mitochondrial genes. SCTransform46 was used to normalize and scale the molecular count data. For Uniform Manifold Approximation and Projection (UMAP) representation and clustering, shared cell populations were matched across samples using Seurat’s integration algorithm for SCTransformed data with reciprocal PCA to identify anchors. Differentially expressed genes between the clusters resulting from Louvain clustering were identified with the FindMarker function based on the SCTransform normalized data, and sorted by fold-change.
ScRNAseq data from the developing mouse embryo was obtained from two publications: Raw counts of the E3.5 to E8.75 embryo dataset from Nowotschin et al.14, including cell type annotations were downloaded from https://endoderm-explorer.com. For visualization, we did not differentiate between the different types of gut tube cells annotated by Nowotschin et al.14, but used “gut tube” as a single label for all these cells. Similarly, we did not differentiate between different samples collected from E8.75 embryos, but pooled these groups with a single E8.75 label. This dataset was integrated with all single-cell transcriptomes from our study in SCANPY47, using log1p-transformed counts after normalization of our data to 10,000 reads per cell. The asymmetric integration and label transfer was performed with ingest and cell type proportions were visualized in R using a custom heatmap function based on pheatmap.
ScRNAseq data and annotations of an embryo dataset focused on AVE development was obtained from the authors15. Integration of this dataset was performed with BELA cells from clusters 3 and 4 in Fig. 3a only, using the same pipeline as for the Nowotschin dataset.
Cell-cell communication analysis
For the inference of cell-cell communication events from scRNAseq data we used LIANA, a LIgand-receptor ANalysis frAmework18. To identify cell-cell communication events in BELAs, we only used transcriptomes from this sample, and grouped them into two lineages according to the clustering in Fig. 3a: All cells from clusters 1 and 2 were grouped as Epi, and cells from clusters 3 and 4 were grouped as VE. The consensus database for ligand-receptor interactions was matched to its mouse ortholog genes using the omnipath database, and interactions were ordered by their consensus rank obtained from LIANA. For Fig. 4a, the top 20 interactions were displayed as an undirected adjacency graph.
Statistics and reproducibility
Quantitative data are represented as mean ± SD. n in figure legends refers to number of biological replicates if not indicated otherwise. Where representative examples such as micrographs are shown, the number of independent experiments and number of structures imaged is indicated in the figure legends. For numbers of independent samples analyzed for each timepoint and condition in Figs. 3m and 4c please refer to the Source Data file. For flow cytometry experiments in Figs. 4 and 5 and Supplementary Figs. 10 and 11, at least n = 20,000 cells were analyzed for each condition in each biological replicate. Statistical analysis was performed in R or in GraphPad Prism 8 (v8.4.3), using unpaired or paired ratio t-tests as indicated in the figure legends. The significance of differential gene expression between clusters in scRNAseq data was assessed with a Wilcox likelihood-ratio test in R.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-024-49380-0