Plant materials and growth conditions
All the plant materials were used in Col-0 or Ler background as indicated. The da1-1, da1-1Ler, da1-ko1 dar1-1, DA1COM, 35S:GFP-DA1, wus-7 and pWUS:WUS-GFP lines were used in previous studies35,36,41,49,56,57,58. The seeds of clv1-1 (NW45), clv2-1 (NW46), clv3-2 (N8066) and wus-1 (N15), were obtained from the Nottingham Arabidopsis Stock Centre (NASC). The da1-1Ler clv1-1, da1-1Ler clv2-1, da1-1Ler clv3-2, da1-1Ler wus-1 and da1-1Ler wus-7 double mutants were generated by crossing da1-1Ler with clv1-1, clv2-1, clv3-2, wus-1 and wus-7, respectively. The pWUS:WUS-GFP; da1-1 was generated by crossing da1-1 with pWUS:WUS-GFP. The pWUS:WUS-GFP; MYC-DA1-PER8 was generated by crossing pWUS:WUS-GFP with MYC-DA1-PER8. All these mutants and double mutants were identified by allele-specific genotyping, transgenic antibiotic resistance screening or GFP signal detection. The primers used above were listed in Supplementary Data 1.
Seeds were sterilized in 100% isopropanol for 1 min and then 5%(v/v) sodium hypochlorite for 10 min. After sufficiently washed with sterile water, seeds were dispersed on 1/2 MS medium containing 1% (w/v) Suc with 0.9% agar and placed at 4°C for 3d. For long-day condition, plants were grown in controlled environmental chamber or in green house with 16 h light /8 h dark. For β -estradiol treatment, plants were grown for 7 days in 1/2MS solid medium and transferred to 1/2MS solid medium with 20 μM β -estradiol for 12 hours.
Confocal microscope observation
For SAM fluorescence signals detection, the shoot apices were detached and prepared from pWUS:WUS-GFP, pWUS:WUS-GFP; da1-1, and pWUS:WUS-GFP; MYC-DA1-PER8 grown under long day condition (16 h light/ 8 h dark) according to a previous study7. Samples were observed using a Zeiss LSM 980 confocal microscopy, and the images used for fluorescencing cells counting were taken by meristem confocal sections at suitable location. All the cell numbers with fluorescence signaling were then quantified by Image J.
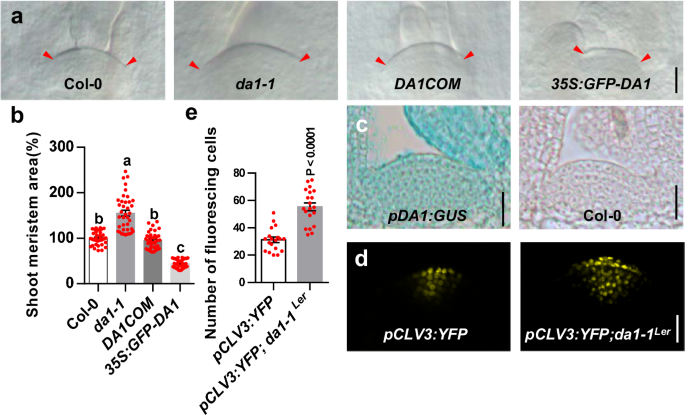
Differential interference contrast microscope
For shoot meristem size observation, 6-d-old and 9-d-old plants were harvested and cleared in clearing solution (Chloral hydrate 80 g, water 30 ml, Glycerol 10 ml) for 12 hrs. Samples were observed with DIC microscope (Leica DM2500, Germany) and photographed using cooled CCD digital imaging system (Olympus BH2, Japan).
SAM size measurement
For SAM size measurement, the measurement range takes L1 layer as the boundary and the junction of SAM and primordia on both sides as the starting points. The details can be found in Supplementary Fig. 3. SAMs were measured using Image J.
Scanning electron microscopy
Samples were fixed in FAA solution with vacuum treatment for 30 min. Fixed samples were dehydrated with a gradual ethanol as 70%, 85%, 95%, and 100% (v/ v), then dried by critical-point drying (Hitachi HCP-2), and subsequently coated with a gold layer. The sputter-coated samples were ready for observation and imaging using scanning electron microscope (Hitachi S-3000N).
Quantitative real-time PCR analysis
6-d-old aboveground part of Col-0 and da1-1 were ground in liquid nitrogen, and the RNAprep pure kit was used to extract total RNA following the manufacturer’s instructions (TIANGEN, Cat. No: DP439-H). cDNAs were synthesized using the FastQuant RT Super Mix kit (TIANGEN, Cat. No: KR108). qPCR reactions were assayed on a Lightcycler 480 machine (Roche Applied Science, USA) using 2×SYBR Green supermix kit (Vazyme, China). All individual reactions were done by three biological replicates, and the data were normalized to the ACTIN2 gene. All primers were listed in Supplementary Data 1.
Yeast two-hybrid assay
The Matchmaker Gold Yeast Two-Hybrid System was employed to perform the yeast two-hybrid assay in yeast strain AH109. AD-DA1 and DA1-LIM + C–BD were used in previous studies4. The CDSs of WUS amplified using primers WUS-BD-F/R (Supplementary Data 1) was inserted into pGBKT7 vector (NcoI and PstI digestion) to get BD-WUS constructs using GBclonart Seamless Clone Kit (GB2001-48, Genebank Biosciences). The CDSs of WUS-HD, WUS-DD, WUS-C and WUS-C2 were inserted into pGADT7 vector (ECOR1 and SAC1 digestion) to get AD-WUS-HD, AD-WUS-DD, AD-WUS-C, and AD-WUS-C2 constructs. Primers are listed in the Supplementary Data 1. The Different combinations of plasmids as indicated were co-transformed in AH109 and grown on SD/-Trp/-Leu plates for 3 days. The protein interactions were then selected by spotting the yeast on SD medium with minus Trp/Leu/His/Ade and grown for 3 or more days.
In vivo co-immunoprecipitation
The genomic sequence of WUS was inserted into PMDC32 (in which the 35 S promotor was cleaved) to generate pWUS:WUS-HA. The construct primers are listed in the supplemental Data 1. The respective transgenic Arabidopsis plants of 35 S:GFP-DA1 and pWUS:WUS-HA were obtained by agrobacterium tumefaciens-mediated transformation. pWUS:WUS-HA; 35S:GFP-DA1; and pWUS:WUS-HA; 35S:GFP plants came from crossing pWUS-WUS-HA with 35S:GFP-DA1 and 35S:GFP, respectively.
Aboveground part of pWUS:WUS-HA;35 S:GFP-DA1, and pWUS:WUS-HA;35S:GFP 10-d-old plants were ground in liquid nitrogen. Total proteins were isolated with buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 5% glycerol, 1 mM EDTA, 1× Roche protease inhibitor cocktail) and incubated with GFP-Trap®_A agarose beads (Chromotek, Cat. No: gta-20) for 1 h with agitation at 4 °C. Beads were washed four times with wash buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 10% glycerol, 0.1% TritonX-100, 1 mM EDTA, and protease inhibitor cocktail). After adding 1×SDS loading buffer, the beads were heated at 98 °C for 5 min, and the corresponding proteins were subjected to sepatate in a 10% or 8% (w/ v) SDS-polyacrylamide gel, and detected with anti-GFP antibody (Abmart, Cat. No: M20004, dilution, 1:5000) and anti-HA antibody (Cwbio, Cat. No: 01239/34220, dilution, 1:5000).
In vitro Pull-down assay
All the constructs were made by infusion cloning kit (GBclonart Seamless Clone Kit, GB2001-48, Genebank Biosciences). The CDS of WUS was infused into pMALC2-MBP digested with SalI and HindIII to generate the MBP-WUS. The CDSs of DA1 was cloned into the pGEX4T-1 vector digested with ECORI and SALI to generate the GST-DA1. Plasmids were transferred into E. coli BL21 (DE3) cells to expression the tag fused proteins. All proteins were induced with 0.4 mM Isopropyl β -D-1-thiogalactopyranoside (IPTG) at different conditions depending on proteins. The bacterial cells were then resuspended with TGH buffer (50 mM HEPES (pH 7.5), 1 mM EGTA, 150 mM NaCl, 1% (v/v) Triton X-100 and 10%(v/v) glycerol). The bacterial suspensions were sonicated on ice for 3 min at 20 amplitudes and centrifugated at 12,000×g for 10 min. The different combinations of MBP-WUS and GST-DA1 proteins with MBP agarose beads (NEB, Cat. No: E8037s) were incubated for 1 h at 4 °C with agitation, respectively. Beads were washed 5 times with washing buffer (50 mM HEPES (pH 7.5), 1 mM EGTA, 150 mM NaCl, 0.5% (v/v) TritonX-100, 1 mM PMSF and 10% (v/v) glycerol). After adding 1×SDS-loading buffer, the beads were heated 5 min at 98 °C and separated by SDS-PAGE gel. The immunoprecipitates were detected by GST antibody (Abmart, Cat. No: M20007M, 1:5000) and MBP antibody (NEB, Cat. No: #E8032, 1:10,000).
Bimolecular fluorescence complementation assay
The CDS of DA1 was amplified by specific primers YN-DA1-F/R, fused with the N-terminal fragment of YFP (nYFP), and subcloned into the linearized pGWB414 vector (digested with XbaI and SalI) using in-fusion enzyme (Genebank Biosciences). The CDS of WUS was amplified by specific primers YC-WUS-F/R, fused with the C-terminal fragment of YFP (cYFP), and subcloned into the linearized pGWB414 vector (digested with XbaI and SalI) using in-fusion enzyme (Genebank Biosciences). Primers used are listed in the supplemental information (Supplementary Data 1). Different combinations of Agrobacterium GV3101 containing the above plasmids were cotransformed into N. benthamiana leaves. After 48 h, YFP fluorescence was observed in leaves using an LSM710 confocal laser scanning microscope (Zeiss)
Cleavage of WUS
The 35 S:HA-DA1 and 35 S:HA-DA1pep constructs were used in previous studies35,59. The cDNA sequences of WUS was inserted into pW1211 and pW1266 to generate 35 S:WUS-FLAG and 35 S:FLAG-WUS by gateway cloning (Invitrogen), respectively. Primers are listed in the Supplementary Data 1.
The rosettes of da1-kol dar1-1 were harvested before bolting to prepare protoplast. The ~0.5 mm fragments of leaves were incubated in enzyme solution (0.3% Macerozyme R-10, 10 mM CaCl2, 0.4 M mannitol, 1.25% Cellulose RS, 20 mM MES at pH 5.7, 5 mM β-Mercatoethanol and 0.1% BSA) for 4 hrs in dark with gentle agitation. After digestion, the W5 solution (154 mM NaCl, 5 mM KCl, 2 mM MES pH 5.7 and 125 mM CaCl2) was added, and followed 10–30 s with strongly shaking. By filtering through 40 μm nylon mesh with three to five washes using W5 solution, the protoplasts were collected by centrifugation at 100 × g for 5 min. The protoplasts were washed twice with W5 buffer and resuspended in MMG solution (0.4 M mannitol, 15 mM MgCl2, and 4 mM MES pH 5.7) around 2× 106 cells mL−1. The 35S:HA-DA1, 35S:HA-DA1pep, 35S:WUS-FLAG, and 35S:FLAG-WUS plasmids were extracted with Plasmid Maxprep Kit (Vigorous, Cat. No: N001). The combinations of constructs (10 μg) were added into 300 μL protoplasts with freshly prepared 330 μL polyethylene glycol (PEG) solution (0.1 M CaCl2, 0.4 M mannitol, 40% w/v PEG4000), and incubated for 20 min on bench. The transfected protoplasts were then resuspended in W5 solution and incubated for 12-16 hrs at 28 °C in the dark. Total proteins were isolated with extraction buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Triton, 0.2% NP-40, 5 mM EDTA, and protease inhibitor cocktail). Proteins were detected by western blots with antibody against FLAG (Abmart, Cat. No: M20008, 1:5000) and HA (Cwbio, Cat. No: 01239/34220, dilution, 1:5000).
The effect of 6-BA treatment on meristem size
For 6-benzylaminopurine (6-BA) treatment, 100 μM stock solutions of 6-BA (Sigma-Aldrich) in ddH2O were dissolved in 1/2MS medium containing 1% (w/v) Suc with 0.9% agar prewarmed at 50 °C to a final concentration of 100 nM. For SAM size measurement, seedlings were grown on 1/2MS solid medium in long-day conditions (16 h light and 8 h dark at 22 °C) for 3 days, then transferred to 1/2MS solid medium with 100 nM 6-BA for 3 days or 8.67 days after seed stratification and then transferred to 1/2MS solid medium with 100 nM 6-BA for 8 h.
Statistics analysis
All data are shown as the mean ± s.e.m. unless indicated otherwise. Statistical analysis was performed using GraphPad Prism 7 software (GraphPad Software, Inc. San Diego, CA, USA). All details on statistics have been indicated in figure legends. The exact P-value was included in the figures. No statistical method was used to predetermine sample size. Images were analyzed with ImageJ.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-024-48361-7