Culture of mammalian cell models
Immortalized Npc1+/+ and Npc1-/- MEFs (gift from Peter Lobel) [21], p62-luciferase expressing Npc1-/- MEFs [29], Atg5+/+ and Atg5-/- MEFs (gift from Noboru Mizushima) [56] and primary Npc1WT and Npc1I1061T (gift from Daniel Ory) [24] MEFs were maintained in DMEM supplemented with 10% foetal bovine serum (FBS), 100 U/mL penicillin/streptomycin and 2 mM L-glutamine (all from Sigma-Aldrich) at 37 °C, and 5% CO2 in a humidified incubator. 293FT cells (Invitrogen, R70007) were cultured as above in a medium supplemented with 1X MEM non-essential amino acids (Gibco). 293GPG cells (gift from Daniel Ory) were cultured as above in a medium supplemented with 1X MEM non-essential amino acid solution, 1 μg/mL tetracycline (Sigma-Aldrich), 2 µg/mL puromycin (Gibco), and 0.3 mg/mL G418 (Gibco) (referred to as 293GPG medium). Human primary fibroblasts derived from control (10263, 10632, 10763, gift from Devin Oglesbee) and NPC1 patient (GM18387, GM18402, GM18417, obtained from Coriell Cell Repositories) were maintained as above in medium supplemented with 15% FBS. Cell authentication was not performed. Mycoplasma contamination was routinely tested using MycoAlert assay kit (Lonza).
iPSC culture and neuronal differentiation
Human induced pluripotent stem cell (iPSC) lines, control_#13 (WIBR-IPS-NPC11920delG/wt, clone #13) and NPC1-2_#26 (WIBR-IPS-NPC1P237S/I1061T, clone #26) iPSC lines were cultured as previously described [20, 57]. The human iPSC lines were cultured on inactivated mouse embryonic fibroblast (MEF) feeder layer in hESC medium comprised of DMEM/F-12, 5% KnockOut Serum Replacement, 1% L-glutamine, 1% non-essential amino acids, 1% penicillin/streptomycin, 4 ng/mL human recombinant basic fibroblast growth factor (bFGF) (all from Gibco), 15% FBS (HyClone) and 0.1 mM β-mercaptoethanol (Sigma-Aldrich) and maintained in a humidified incubator with 5% CO2 and 5% O2 at 37 °C. For experimentation, the iPSCs were cultured feeder-free on Geltrex basement membrane matrix in StemFlex Basal Medium supplemented with StemFlex 10X Supplement (all from Gibco).
Neural stem cells (NSCs) were differentiated from iPSCs, as described previously [58, 59]. NSCs were cultured on Poly-L-ornithine and Laminin (PO-L) (Sigma-Aldrich) coated plates or flasks in N2B27 medium comprised of DMEM/F-12 and Neurobasal medium in 1:1 ratio, 1% N-2 supplement, 2% B-27 supplement, 1% penicillin/streptomycin (all from Gibco), 0.1% β-mercaptoethanol (Sigma-Aldrich) supplemented with 10 ng/mL FGF-2 (Miltenyi Biotec) and 10 ng/mL EGF (PeproTech), and were maintained in a humidified incubator with 5% CO2 at 37 °C. NSCs were passaged twice a week with 0.05% Trypsin-EDTA (Gibco), and the medium was changed on alternate days.
Neuronal differentiation of human iPSC-derived NSCs was carried out as described previously [58, 59]. The NSCs were seeded as above on PO-L coated plates in N2B27 medium without FGF-2 and EGF. At day 4 of neuronal differentiation, cells were treated with 10 µM DAPT (Tocris) to prevent cell proliferation. The N2B27 medium (without FGF-2 and EGF) was changed every 2 days and neuronal differentiation was carried out for 4 weeks. The neurons generated in vitro were cortical in nature [58, 59].
The control and NPC1 patient-derived iPSCs were originally generated in the lab of Rudolf Jaenisch at the Whitehead Institute for Biomedical Research. These cell lines were used for this study in the lab of Sovan Sarkar at the University of Birmingham under material transfer agreements, UBMTA 15-0593 and UBMTA 15-0594. All experiments were performed in accordance with ISSCR and institutional guidelines and regulations.
Generation of stable cell lines
Re-introduction of the NPC1 gene into Npc1-/- MEFs was achieved by retroviral transduction. For retrovirus production, 293GPG cells were seeded in a 10 cm dish (5.5 × 106 cells/10 mL/dish) 48 h prior to DNA transfection. At 90% confluency, the culture medium was replaced with tetracycline-free 293GPG medium and transfected with either empty or NPC1 retroviral expression plasmid (gift from Daniel Ory) with 4 μg of DNA using Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer instruction. Following overnight incubation, the medium was replaced with 293GPG medium containing 20% FBS. Virus-containing medium was collected 48 h post-transfection and was filtered through a 0.45 μm membrane filter and overlaid on 70% confluent cells in the presence of 10 μg/mL polybrene (Sigma-Aldrich).
Generation of cells stably expressing YFP-Parkin, mt-mKeima, or Halo-GFP-LC3B was achieved by packaging retroviruses in 293FT cells. Cells were seeded in a 10 cm dish (6.0 × 106 cells/10 mL/dish) in antibiotic-free culture medium. Next day, cells were transfected with plasmids containing the packaging gag/pol and envelope pCMV-VSV-G genes, and the YFP-Parkin-IRES-zeo (gift from Douglas Green and Stephen Tait, Addgene, 61728) [60], pCHAC-mt-mKeima (gift from Richard Youle, Addgene, 72342) [61] or pMRX-No-HaloTag7-mGFP-LC3B (gift from Noboru Mizushima, Addgene, 184901) [35] using Lipofectamine 2000. Following overnight transfection, the medium was replaced with a fresh antibiotic-free medium that was collected after 24 h. Virus-containing medium was collected and used for transduction as above.
Galactose medium culture and supplementation
To induce mitochondrial respiration in MEFs and human primary fibroblasts, cells were cultured in galactose medium (glucose-free DMEM (Gibco) supplemented with 10 mM D-galactose, 10 mM HEPES, 1 mM sodium pyruvate, 4 mM L-glutamine, 100 U/mL penicillin/streptomycin and 10% FBS (all from Sigma-Aldrich) 24 h post seeding. Galactose medium was supplemented with various compounds as indicated in figure legends: 100 nM bafilomycin A1 (BafA1, Enzo Life Sciences, BML-CM110-0100), 10 µM celecoxib (Cele, Sigma-Aldrich, SML3031), 30 µM memantine (Mem, Sigma-Aldrich, M9292), 5 mM nicotinamide (NAM, Sigma-Aldrich, N0636), 2 mM nicotinamide riboside (NR, ChromaDex), 100 or 300 µM reduced nicotinamide riboside (NRH) [36] and 50 µM nicotinic acid riboside (NAR) [36]. All compound supplements were added at 0 h.
Screening of autophagy inducers
p62-luciferase expressing Npc1-/- MEFs were plated into white walled 384 well plates (0.2 × 104 cells/20 µL/well). Cells were then cultured in DMEM supplemented with or without 1 µg/mL doxycycline (Sigma-Aldrich) for 24 h. After three washes with PBS, cells were treated with 935 compounds (10 µM) from an FDA-approved drug library (LifeArc) for 24 h. Following the treatment, 5 µL of Bright-Glo (Promega) was added to each well, and luminescence was assessed on a BMG Omega POLARstar plate reader. Luminescence fold change (treated to control) was calculated using adjusted luminescence signals by subtracting background signals (-Dox datasets), Statistical significance of deviation of a given point from an unconstrained linear regression model comparing to +Dox with –Dox datasets was defined as P values calculated by t-test and adjusted by Benjamini-Hochberg FDR correction.
MS-based metabolomics
Metabolite extraction for liquid-chromatography-mass spectroscopy (LC-MS) was performed on MEFs and human primary fibroblasts cultured in galactose medium for 48 h or 7 d, respectively. Cells were washed once with cold PBS (CST) and lysed at a concentration of 2 × 106 cells/mL in a metabolite extraction buffer (50% methanol (Fisher Scientific), 30% acetonitrile (Sigma), 20% dH2O). Samples were vortexed for 45 s, centrifuged at 16,100 g and supernatants were subjected to LC-MS as follows, using a three-point calibration curve with universally labeled carbon-13/nitrogen-15 amino acids for quantification. Prepared samples were analysed on an LC-MS platform consisting of an Accela 600 LC system and an Exactive mass spectrometer (Thermo Scientific). A Sequant ZIC-pHILIC column (4.6 mm x 150 mm, 5 µm) (Merck) was used to separate the metabolites with the mobile phase mixed by A = 20 mM ammonium carbonate in water and B=acetonitrile. A gradient program starting at 20% of A and linearly increasing to 80% at 30 min was used followed by washing (92% of A for 5 mins) and re-equilibration (20% of A for 10 min) steps. The total run time of the method was 45 min. The LC stream was desolvated and ionised in the HESI probe. The Exactive mass spectrometer was operated in full scan mode over a mass range of 70–1,200 m/z at a resolution of 50,000 with polarity switching. The LC-MS raw data was converted into mzML files by using ProteoWizard and imported to MZMine 2.10 for peak extraction and sample alignment. A house-made database integrating KEGG, HMDB, and LIPID MAPS was used for the assignment of LCMS signals by searching the accurate mass and the metabolites used in the manuscript were confirmed by running their commercial standards. Finally, peak areas were used for comparative quantification. Output from MS-based metabolomics was subjected to statistical analysis by MetaboAnalyst 5.0. The variables were normalised by auto-scaling (mean-centered and divided by SD of each variable) by the MetaboAnalyst platform and then subjected to principal component analysis (PCA), heatmap and hierarchical clustering analysis with Euclidean distance, and complete-linkage method. Statistical significance was determined using the Student’s t-test corrected with the false discovery rate (FDR) method of Benjamini and Hochberg.
Seahorse assay
MEFs seeded into Seahorse XF24 V7 assay plates (0.8 × 104 cells/well) (Agilent Technologies, 100777-004) were cultured in glucose medium for 20 h. The media was switched to unbuffered glucose medium (DMEM (Sigma-Aldrich, D5030), 3% FBS, 5 mM glucose, 1 mM sodium pyruvate, 2 mM L-glutamine, 1 mM HEPES, pH 7.4) 1 h before the assay, and the plate was incubated at 37 °C without CO2. Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were determined using Seahorse XF24 analyzer (Agilent Technologies) in the presence of different respiratory and glycolysis inhibitors which were sequentially added as follows: 1.5 µM oligomycin, 3 µM Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and a mixture of 0.5 µM Rotenone, 2.5 µM Antimycin A, and 50 mM 2-deoxyglucose. Cellular energetics for ATP production rates by mitochondrial oxidative phosphorylation and glycolysis were calculated by using the OCR and ECAR based on the methods described [62, 63], taking into account the acidification rates due to mitochondrial CO2 production, and were adjusted to protein concentration determined by the DC protein assay (BioRad). To measure CI- and CII-linked respiration, cells were permeabilised using Seahorse XF Plasma Membrane Permeabilizer (Agilent Technologies), and OCR was measured in the assay buffer (115 mM KCl, 10 mM KH2PO4, 2 mM MgCl2, 3 mM HEPES, 1 mM EGTA and 0.2% fatty acid-free BSA, pH 7.2, at 37 °C) with CI substrates (10 mM pyruvate and 1 mM malate) or CII substrate (4 mM succinate and 0.5 µM rotenone). During analysis, the following compounds were added to test mitochondrial activity and cellular bioenergetics flux: 4 mM ADP, 0.5 µM oligomycin, 2.5 µM FCCP, and 2.5 µM antimycin A.
To measure OCR in galactose culture, MEFs were seeded into Seahorse xFe96/XF Pro assay plates (0.8 × 104 cells/well) (Agilent Technologies, 103794-100). Following 20 h culture in galactose medium, the media was switched to unbuffered galactose medium (DMEM (Sigma, D5030) 3% FBS, 10 mM Galactose, 1 mM Pyruvate, 2 mM L-Glutamine, 1 mM HEPES, pH 7.4), and the plate was incubated at 37 °C without CO2 for 1 h. OCR was determined using the Seahorse XFe96 analyzer (Agilent Technologies). During analysis, the following respiratory inhibitors were added sequentially: 2 µM oligomycin, 2 µM FCCP, and a mixture of 0.5 µM rotenone and 2.5 µM antimycin A. OCR data was normalized to cell content/well by staining cells with 0.2% crystal violet. Stained cells were lysed with 1% SDS, and the absorbance of the resulting solution was measured at 595 nm using a microplate reader (FLUOstar Omega, BMG Labtech).
Electron Microscopy
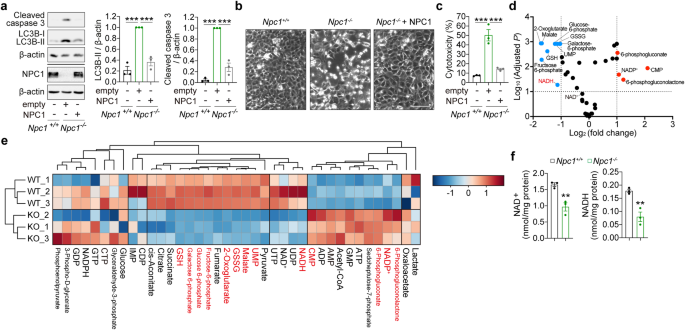
MEFs seeded in a 6-well plate (0.3 × 106 cells/2 mL/well) were cultured in galactose medium for 20 h. Cells were trypsinised, washed, collected and fixed overnight in 2% glutaraldehyde in 0.1 M cacodylate buffer. After rinsing in buffer, the cells were post-fixed in 1% osmium tetroxide + 1.5% potassium ferricyanide, rinsed in deionized water then dehydrated through a graded series of acetone. Cells were infiltrated with epoxy resin (TAAB medium) and polymerized at 60 °C for 36 h. Ultrathin sections (70 nm) were picked up on copper grids and stained with uranyl acetate and lead citrate before being viewed on a 100 kV CM100 TEM (FEI). Images of 10 cells per cell line were collected and quantified. Mitochondrial morphology in a slice was scored as either ‘normal’ or ‘abnormal’ (swollen or disputed cristae structure) and expressed as a ratio of mitochondria with ‘abnormal’ morphology per cell.
NAD+ and NADH measurements
Measurements of NAD+ and NADH in mammalian whole-cell lysates, and in brain and liver mouse tissues were performed as described in a published protocol [8, 64]. NAD+ or NADH was extracted from 4 × 106 cells (NAD+) or 8 × 106 cells (NADH) cells by probe sonication with an acidic solution (10% trichloroacetic acid (TCA) (Sigma-Aldrich) or basic solution (0.5 M sodium hydroxide (Sigma-Aldrich), 5 mM EDTA (Sigma-Aldrich)) respectively. NADH samples were heated at 60 °C for 30 min. Samples were centrifuged at 16,100 g for 3 min at 4 °C. Supernatants were collected and 10% volume of 1 M Tris (Sigma-Aldrich) was added to adjust pH, followed by NAD+ and NADH measurements. A small amount of supernatant (NADH) or the pellet (NAD+) resolved in 0.2 M sodium hydroxide were used to measure protein concentrations. NAD+ and NADH levels were determined by the fluorescence intensity of resorufin produced by an enzymatic cycling reaction using resazurin, riboflavin 5′-monophosphate, alcohol dehydrogenase, and diaphorase (all from Sigma-Aldrich). Fluorescence intensity was monitored every minute for a total 60 min using a microplate reader (FLUOstar Omega, BMG Labtech). NAD+ and NADH levels were determined by a β-NAD (Sigma-Aldrich) standard curve and adjusted to protein concentration determined by the DC protein assay (BioRad).
Image acquisition
Fluorescence images were obtained using an inverted DMi8 microscope (Leica) with a Plan-Apochromat 63x/1.40 oil immersion lens, equipped with an ORCA-Flash4v2.0 camera (Hamamatsu) (MitoSOX and mt-mKeima imaging), an Axio observer Z1 microscope (Zeiss), with a Plan-Apochromat 20x/0.8 M27 air immersion objective, equipped with an Axiocam 503 camera (LC3B staining in human primary fibroblasts), EVOS FL Cell Imaging System (Thermo Fisher Scientific) with AMG 10x Plan FL and AMG 40x Plan FL lens (iPSC experiments), or an LSM700 microscope (Zeiss) with a C-Apochromat 40x/1.20 water immersion lens (mitochondrial ΔΨm measurements in MEFs). Deconvolved images were generated using Huygens Essential software (version 20.10, Scientific Volume Imaging). Images were analyzed in Fiji/ImageJ (version 1.53c; NIH), and quantification was performed on at least 50 cells per condition.
ROS measurement
Cells seeded in a 35 mm glass bottom dish (MatTek) were co-stained with 2.5 μM mitoSOX (Invitrogen, M36008) and 100 nM MitoTracker Green (Invitrogen, M7514) for 10 min and washed three times with galactose medium. Fluorescence images were obtained described as above. Fluorescence intensity was analysed as outlining single cells as regions of interest and calculation of the raw integrated density value per cell.
For the flow cytometry-based ROS analysis, human primary fibroblasts were cultured in galactose medium for 7 d, seeded in 6-well plate (0.15 × 106 cells/well) and cultured in galactose medium for 48 h. Cells were stained with 10 µM CM-H2DCFDA (Invitrogen, C6827) for 30 min in the dark at 37 °C. Cells were then trypsinized and re-suspended in FACS sorting medium (3% FBS (BioSera) in PBS). Flow cytometry data (10,000 counts) was acquired on BD FACSCanto™ benchtop analyser (BD Biosciences), and analysed using the FlowJo software.
Mitochondrial ΔΨm measurements
MEFs were grown in a 96-well glass bottom plate (Greiner Bio-One) (0.5 × 104 /100 μL/well, 24 h). Following culture in galactose medium for 68 h, cells were co-stained with 16.7 nM tetramethylrhodamine methyl ester (TMRM; Invitrogen, T668) and 100 nM Mitotracker Green (MTG) for 30 min in conditioned galactose medium (68 h culture on Npc1-/- MEFs, collected and filtered through a 0.22 µm pore-size filter). Live cell imaging was performed in a maintained atmosphere of 37 °C and 5% CO2. TMRM and MTG raw integrated density values per cell were quantified by outlining single cells as regions of interest. Mitochondrial ΔΨ was expressed as a ratio of TMRM to MTG. Quantification was performed on at least 30 cells per condition.
Measurements of ΔΨm in iPSC-derived cortical neurons were performed with TMRE (Invitrogen, T669) staining [65]. Cells were loaded with Microscopy Medium comprising 120 mM NaCl, 3.5 mM KCl, 0.4 mM KH2PO4, 5 mM NaHCO3, 1.2 mM NaSO4, 20 mM HEPES and 15 mM glucose in dH2O adjusted to pH 7.4 and supplemented with 1 mM CaCl2 (all from Sigma-Aldrich), and incubated with 500 nM TMRE for 1 h at 37 °C. The fluorescence signals of TMRE was acquired using EnSpire Multimode microplate reader (PerkinElmer) for a period of 5 min to get basal fluorescence, and again for TMRE for another 5 min after the addition of 10 μM FCCP (fluorocarbonyl cyanide phenylhydrazone). The baseline fluorescence was calculated as the mean of the last 5 fluorescence readings before the addition of FCCP, and the delta (Δ) fluorescence was calculated by subtracting the basal fluorescence from the average of first 5 fluorescence readings after FCCP treatment. Data were obtained as relative fluorescence units, normalised to protein concentration via Bio-Rad Protein Assay (Bio-Rad), and expressed as a percentage of the control condition.
Immunoblotting
Immunoblotting was performed as described previously [66]. In brief, cells were lysed in RIPA buffer (Sigma-Aldrich) supplemented with 1× Halt™ protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentration was measured using DC Protein Assay (Bio-Rad), and samples were prepared by boiling in Laemmli sample buffer (Bio-Rad) in the presence of 2.5% β-mercaptoethanol. Equal amounts of protein (20–40 μg) were subjected to SDS‐PAGE and transferred to PVDF membranes. Membranes were first blocked in 5% milk (Merck Millipore) in PBS with 1xTween® 20 (Sigma-Aldrich) for 1 h at room temperature and incubated with primary antibodies overnight at 4 °C. Secondary antibodies conjugated to horseradish peroxidase (HRP) for rabbit (Sigma-Aldrich, A0545) or mouse (Sigma‐Aldrich, A2554) were used at 1:5,000 dilution for 1 h at room temperature. Chemiluminescence detection was achieved using Clarity Western ECL Substrate (Bio-Rad) and a LAS4000 CCD camera system (Fujifilm) or an iBright CL1500 imaging system (Invitrogen). The following primary antibodies were used:
β-actin (St John’s Laboratory, STJ96930, 1:5000), Cleaved caspase-3Asp175 (CST, 9661, 1:250), HaloTag (Promega, G9211, 1:2000), LC3B (CST, 3868, 1:1000), NPC1 (abcam, ab134113, 1:1000), phospho-p70S6KThr389 (CST, 9234, 1:1000), p70S6K (CST, 9202, 1:1000), phospho-ULK1Ser757 (CST, 6888. 1:1000). Densitometry analyses of immunoblots were done using Fiji/ImageJ (version 1.53c; NIH).
Immunofluorescence
Human primary fibroblasts seeded onto 13 mm coverslips were cultured for 48 h. Cells were fixed and permeabilised in −20 °C 100% methanol for 5 min and blocked in 5% normal goat serum in PBS for 1 h and incubated with LC3B antibody (CST, 3868S, 1:1000) overnight at 4 °C. Cells were washed and incubated with Alexa Fluor 488 goat anti-rabbit (H + L) antibody (1:1000; Thermo Fisher Scientific; A-11008) for 1 h at room temperature. Coverslips were mounted on slides with Prolong Gold antifade reagent with DAPI (Invitrogen, P36931).
iPSC-derived cortical neurons were washed in PBS, fixed with 4% formaldehyde at room temperature for 15 min, permeabilised with 0.5% Triton X-100 for 10 min (except LC3B staining) or with pre-chilled methanol for 5 min (for LC3B antibody), and incubated with Blocking Buffer (5% goat or donkey serum (Sigma-Aldrich) in PBS with or without (LC3B staining) 0.05% Tween 20 for 1 h at room temperature. Cells were then incubated overnight with primary antibodies at 4 °C, followed by incubation with Alexa Fluor conjugated secondary antibodies for rabbit (Thermo Fisher Scientific, A21206) or mouse (Thermo Fisher Scientific, A21203) for 1 h at room temperature. The coverslips were mounted on glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen). The following primary antibodies were used: rabbit α-LC3B (Novus Biologicals, NB100-2220, 1:200), mouse α-p62 (BD Biosciences, 610832, 1:200), rabbit α-MAP2 (CST, 8707, 1:200) mouse α-TUJ1 (CST, 4466, 1:200).
Cell death assays
Adherent and floating MEFs were collected and processed by protein extraction and immunoblot analysis at 24 h (Atg5+/+ and Atg5-/- MEFs) and 72 h (Npc1+/+ and Npc1−/− MEFs) after media switch. Representative phase-contrast images were obtained on an inverted DM-IL Leica microscope equipped with an Invenio 3SII digital camera (3.0 Mpix Colour CMOS; Indigo Scientific).
Cytotoxicity was measured using Cytotox-Glo Cytotoxicity Assay (Promega, G9291) according to manufacturer instruction. Cells in a 96-well white plate (Greiner) cultured for the indicated times after media switch were incubated with Cytotox-Glo Assay Reagent for 15 min at room temperature in the dark, then luminescence was measured using a GloMax plate-reader (Promega) or EnSpire Multimode plate reader (PerkinElmer) and the readings obtained were attributed to the basal cytotoxicity per well (first reading). To estimate cell population per well, cells were further incubated with Lysis Reagent for 30 min at room temperature in the dark, after which luminescence was measured again (second reading). Cytotoxicity data were normalised by dividing the first reading (basal cytotoxicity per well) to the second reading (indicative of cell population per well) and expressed as a percentage.
TUNEL staining for apoptotic neuronal cells were performed using Click-iT Plus TUNEL Assay kit (Invitrogen, C10617), according to the manufacturer’s protocol. For detection of TUNEL+ apoptotic nuclei specifically in neurons, cells were subjected to immunofluorescence by blocking with 3% BSA (in PBS) followed by incubation with anti-TUJ1 antibody (BioLegend, 801201) overnight at 4 °C, and thereafter incubated with Alexa Fluor 594 secondary antibody for 1 h at room temperature. Coverslips were mounted on glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen). The quantification of TUNEL+ apoptotic nuclei in TUJ1+ neuronal cells was performed via fluorescence microscopy, as previously described [20]. The percentage of TUNEL+ nuclei was calculated from the total number of TUJ1+ cells analysed.
Assessment of autophagy
LC3B turnover assay was performed to monitor autophagy flux in MEFs and human primary fibroblasts[34]. Cells were cultured in galactose medium in the presence or absence of BafA1 (to block lysosomal degradation) for 24 h, followed by immunoblotting. LC3B-II flux was expressed by subtracting the signals of LC3B in BafA1-untreated conditions from those of LC3B in BafA1-treated conditions. Additionally, LC3B-positive autophagosomes in human primary fibroblasts were visualised by immunofluorescence analysis. The number of LC3B puncta per cell was quantified.
Halo processing assay was conducted to assess autophagy-inducing activities of small molecules [35]. Npc1-/- MEFs expressing Halo-GFP-LC3B were treated with compounds and 20 µM 7-Bromo-1-heptanol (HaloTag-blocking agent) (Thermo Scientific Chemicals, H54762) [67], followed by immunoblot analysis using Halo antibody. Autophagic activity was quantified as a ratio of Halo monomer per Halo-GFP-LC3B.
In iPSC-derived cortical neurons, autophagic activity was assessed by the degradation of autophagy marker proteins, p62 and LC3B, by immunofluorescence. The number of LC3B or p62 puncta in TUJ1- or MAP2-positive cells was quantified.
Mitophagy assay
MEFs expressing YFP-Parkin and human primary fibroblasts were transduced with mt-mKeima as above. Cells were seeded in a 35 mm glass bottom dish, and the live-cell mt-mKeima signal was obtained using a DMi8 microscope (Leica). Mitophagy events were determined as following steps using Fiji/ImageJ (version 1.53c; NIH) [68]. Images were masked by applying MaxEntropy threshold algorithm to the images obtained with 561 nm excitation to remove low red signal and background. Within the masks, signals of mt-mKeima were adjusted by applying Enhanced Contrast plugin with saturated = 0.1, normalise, equalise options. Then, images were generated by subtracting the signal at a 480 nm excitation (reporting neutral pH-environment) from the signal at a 561 nm excitation (reporting an acidic pH-environment). Resulting images were binarised with the MaxEntropy threshold algorithm to extract mitolysosomes. The number of puncta per cell in images was quantified by outlining single cells as regions of interest and counted using Analyze Particles plugin.
Statistical analyses
All experiments were carried out in three or more biological replicates from independent cell culture or cell lines (human primary fibroblasts). Quantifications of data and statistical analysis of metabolomics are described under the corresponding method section. Sample size calculation was not performed. Graphical data denote the mean ± SEM (of n = 3 or more biological replicates or cell lines) and are depicted by column graph scatter dot plot, using Prism 8.4.3 software (GraphPad). Unless indicated otherwise, the P values was determined by Student’s t-test (two-tailed, unpaired) between two groups or by one-way ANOVA followed by multiple comparisons with the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli (with an FDR value of 0.05) using Prism 8.4.3 software (GraphPad). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns (non-significant).
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41419-024-06770-y