Synthesis
The peptidomimetics were synthesized according to a procedure described previously by our team14. The biotin moiety was attached to the N-terminal amino group using a molar excess of Bt-NHS:DIPEA (3:1.5) in NMP. The completeness of the reaction was monitored by the Kaiser test.
Cell lines
The healthy cell line HB2 (human breast epithelial cells) was obtained from Merck (Germany), the cancer cell line MDA-MB-231 (human breast cancer epithelial cells) was obtained from ATCC (United Kingdom), the cancer cell line SKBR3 (human mammary gland cancer) was obtained from CLS (Germany), and the cancer cell line T47D (human breast cancer epithelial cells) was obtained from CLS (Germany). All cell lines were cultured at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) supplemented with 4.5 g/L glucose, 10% fetal bovine serum (FBS) and 1% penicillin‒streptomycin solution (100 units of penicillin and 100 µg/mL streptomycin). The HB2 cell line requires the addition of 5 µg/mL insulin and 5 µg/mL alcoholic hydrocortisone solution.
Complex formation and electrophoresis assay
FITC-Strp: Bt-O2Oc-[Dap(GO2)]6-O2Oc-NH2/Bt-O2Oc-[Dap(GO2)]8-O2Oc-NH2
FITC-streptavidin (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was incubated with increasing concentrations of polymers with N-terminal biotin (compound 1 or 2) or free amino groups (compound 1a or 2a) at molar ratios of 1:1, 1:2, and 1:4 for one hour at RT. Each complex was separated on a native polyacrylamide gel at a concentration of 0.1 or 0.2 µg per lane with polymer and FITC-Strp as controls. A 15-well Native PAGE 4–16%, Bis–Tris, 1.0 mm, Mini Protein Gel (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was used. The separation conditions were as follows: 50 mM Bis–Tris, 50 mM tricine, pH 6.8 (running buffer), and constant voltage of 150 V for 2 h at 24 °C. Loading buffer composition: 200 mM Bis–Tris, 6 N HCl, 200 mM NaCl, 40% w/v glycerol, 0.004% bromophenol blue, pH 7.2. Cathode buffer composition: 0.4% Coomassie G-250 diluted 10 times in double distilled water in running buffer. At the final step, the gel was placed in 100 mL of fix solution (40% methanol and 10% acetic acid in water) and microwaved at 1100 watts for 45 s. Next, the gel was shaken for 30 min at room temperature. The fixing solution was removed, and the whole procedure was repeated. After this, the gel was ready for visualization.
Strp-β-gal complex with Bt-peptidomimetic
The hybrid protein construct streptavidin-β-gal (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at a concentration of 2 mg/mL in H2O was incubated for 1 h at RT in molar ratios of 1:1, 1:2, or 1:4 with compound 1 or 2; nonbiotinylated compounds (1a or 2a) were used as controls. All samples were subjected to electrophoretic separation at a protein concentration equal to 7.5 µg per well.
Acidic native gel
The polyacrylamide gel was cast and run as follows: stacking gel, 4% acrylamide, pH 5.0 (0.22 M acetic acid/KOH buffer); resolving gel, 8% acrylamide, pH 4.0 (0.075 M acetic acid/KOH buffer); running buffer, 3.55% β-alanine/acetic acid in water, pH 4.0; loading buffer, 50% glycerol in 0.22 M acetic acid/KOH buffer, pH 5.0 with 0.0005% crystal violet.
The gel was run with reverse polarization at 5 mA initially (45 min); then, after the samples migrated out from the stacking gel, it was run at 20 mA (2 h). Images of the gel were taken using a Fusion Fx (Vilber Lourmat, France) system.
Cell studies
FITC-streptavidin: Bt-peptidomimetic complex cellular uptake
Labeled FITC-streptavidin (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at a concentration of 0.2 mg/mL in H2O was incubated with peptidomimetic (biotinylated (1 or 2) or lacking biotin (1a or 2a)) at molar ratios of 1:2 and 1:4 for one hour at RT.
All cells were seeded in 24-well plates (initial number of cells: 104) and cultured in 0.5 mL of appropriate medium for 24 h. Next, the cells were washed three times with PBS, and 0.5 mL of peptidomimetic (1, 2 or 1a, 2a) in medium was added. The concentration of the all peptidomimetic (1, 2 or 1a, 2a) was 10 µM (molar ratio 1:2) or 20 µM (molar ratio 1:4) respectively. After the indicated time period (2 h, 4 h or 24 h), the cells were washed with PBS and cultured in FluoroBrite DMEM (Gibco, New York, USA).
An ECLIPSE Ti-E inverted fluorescence microscope (Nikon, Tokyo, Japan) was used to evaluate the complex distribution in the cells. The appropriate filters were used for the detection of DAPI and FITC.
Streptavidin-β-gal: Bt-peptidomimetic complex cellular uptake
Streptavidin-β-gal (Invitrogen by Thermo Fisher Scientific, Waltham, Massachusetts, USA) at a concentration of 0.2 mg/mL in H2O was incubated with peptidomimetic (biotinylated (1,2) or lacking biotin (1a, 2a)) at molar ratios of 1:2 and 1:4 for 1 h at RT.
All cells were seeded in 96-well plates (initial number of cells: 103) and cultured in 0.1 mL of appropriate medium for 48 h. Next, the cells were washed with PBS (three times), and 50 µL of appropriate peptidomimetic (1 or 2) in medium was added. The concentration of each peptidomimetic was 10 µM (molar ratio 1:2) or 20 µM (molar ratio 1:4). Cells were cultured for 24 h and then subjected to PBS washes (2 times). After the selected time period, 0 h (t24h) or 2 h (t24h+2h) (see scheme on Fig. 2) the cells were washed with PBS and lysed with Reporter Lysis Buffer RLB (Promega, Madison, Wisconsin, USA) according to the manufacturer’s manual. All lysates were immediately snap frozen at − 80 °C.
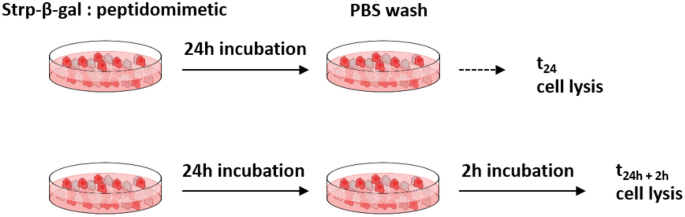
Incubation scheme for Strp-β-gal:peptidomimetics. Additional two hours of culturing allow the membrane bound complex to dissociate.
Activity studies
The enzymatic activity of β-gal was monitored using resorufin β-d-galactopyranoside (Thermo Fisher Scientific, Waltham, Massachusetts, USA) (final concentration = 2.5 mg/mL dimethyl sulfoxide (DMSO)). The increase in fluorescence was monitored in triplicate using a FluoroStar OMEGA microplate reader (BMG LABTECH, Ortenberg, Germany) with extinction and emission wavelengths of 541 nm and 585 nm. Assay conditions were as follows: 2.5 µL of lysate, 100 µL assay buffer (0.1 M phosphate buffer, pH 7.0) and 5 µL of substrate. The assay was run for 120 min at 37 °C.
In-cell β-gal activity detection
Cells were incubated with the formed complexes for 24 h in culture medium washed by PBS and cultured for additional 2 h or directly subjected to the next step. After this, two washes with PBS were applied to each system; then, the cells were fixed in 3.7% paraformaldehyde in PBS for 15 min. Next, the cells were washed twice in PBS, and 125 µL of reaction mixture was added to each system. The reaction buffer consisted of 40 mM citric acid, 40 mM sodium phosphate, 300 mM NaCl, 10 mM mercaptoethanol, and 4 mM MgCl2 (pH 6.0) with 2 mM X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactopyranoside) added immediately prior to use from a 34 mM stock in DMSO17. The plate was incubated for two hours at 37 °C and observed under an Olympus IX51 microscope (Olympus, Tokyo, Japan). Application of the X-Gal reagent in the presence of β-gal activity results in a blue, insoluble product that precipitates and can be monitored under an optical microscope.
Cytotoxicity studies
All cell lines were seeded at a concentration of 5 × 103 per well in 96-well plates and cultured for 48 h in 100 µL of medium. Next, the medium was replaced with new medium containing the target amount of tested compounds or their complexes.
The following compounds were tested: Bt-O2Oc-[Dap(GO2)]6-O2Oc-NH2 (Compound 1), where Bt—biotin, O2Oc—8-amino-3,6-dioxaoctanoic acid, Dap—L-2,3-diaminopropionic acid, GO2—8-amidino-3,6-dioxaoctanoic acid, Bt-O2Oc-[Dap(GO2)]8-O2Oc-NH2 (Compound 2), O2Oc-[Dap(GO2)]6-O2Oc-NH2 (Compound 1a) and O2Oc-[Dap(GO2)]8-O2Oc-NH2 (Compound 2a) at concentrations of 1, 5, 10, 20, 50, and 100 µM in the well;
-
Complex FITC-Strp: Compound 1/Compound 2 and FITC-Strp: Compound 1a/Compound 2a in molar ratios of 1:2 and 1:4 at concentrations of 1, 10, 20, and 30 µM in the well;
-
Complex Strp-β-gal: Compound 1/Compound 2 and complex Strp-β-gal: Compound 1a/Compound 2a in molar ratios of 1:2 and 1:4 at concentrations of 1, 10, and 20 µM in the well.
Each incubation was run in triplicate for 24 h. Untreated cells were used as a control. After 24 h, the medium was removed, and the cells were incubated with MTT-containing medium for 4 h. Next, DMSO was added to each well, and the results were read using a plate reader (SPECTROstar Nano, BMG LABTECH, Ortenberg, Germany) at two different wavelengths, 570 nm and 690 nm.
Immunofluorescence staining and analyses of β-gal in selected cell lines
HB2 or MDA-MB-231 cells were seeded and grown on glass slides (removable 3-well chamber, IBIDI, Gräfelfing, Germany). On the day of the experiment, cells were incubated with a freshly prepared complex of Strp-β-gal and the chosen compound. At the appropriate time points (t24h and t24h+2h), the cells were washed and subjected to further experimental procedures.
After incubation, the cells were fixed for 10 min in 1% paraformaldehyde in PBS, washed in PBS, permeabilized in 1% Triton X-100 in PBS, and blocked for 4 h with blocking buffer consisting of 2% FBS, 2% bovine serum albumin (BSA) and 0.2% fish gelatin in PBS (lacking Ca2+ and Mg2+). Next, the cells were incubated for 1 h with β-galactosidase monoclonal primary antibodies (Thermo Fisher Scientific, Waltham, Massachusetts, USA, cat number MA1-152; 1:50), washed four times in 0.1% Tween in PBS followed by DyLightTM488-conjugated goat anti-mouse IgG secondary antibody diluted in blocking buffer for 45 min (Thermo Fisher Scientific, Waltham, Massachusetts, USA, cat number 35502; 1:50). After 30 min of incubation, Hoechst reagent was added to all systems. After 45 min of incubation, the cells were washed four times in 0.1% Tween in PBS. Blocking solution was used instead of primary antibody as a control for nonspecific staining. Coverslips were mounted on the slides using Fluoromount Aqueous Mounting Medium (Sigma‒Aldrich, Darmstadt, Germany). The specimens were imaged with an ECLIPSE Ti-E confocal laser scanning microscope. To determine the mean fluorescence intensity profile of the immunocomplexes, the mean intensity values for green channels was analyzed using NIS-Elements software (Nikon, Tokyo, Japan) with the Automated Measurement Results option.
Statistical analyses
The statistical analyses were performed using GraphPad Prism 8 (Graphpad software, USA). The Shapiro–Wilk test was used to determine normality of the datasets. For data with a normal distribution, the unpaired t-test was used. For other cases, the nonparametric Mann–Whitney test was applied. The results are expressed as mean ± SEM. Values of p ≤ 0.05 were considered statistically significant.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41598-024-64187-1