PLMSearch pipeline
PLMSearch consists of three steps (Fig. 1a-c). (1) PfamClan. Initially, PfamScan54 identifies the Pfam clan domains of the query protein sequences. Subsequently, PfamClan searches the target dataset for proteins sharing the same Pfam clan domain with the query proteins. In addition, a limited number of query proteins lack any Pfam clan domain, or their Pfam clans differ from any target protein. To prevent such queries from yielding no results, all pairs between such query proteins and target proteins will be retained. (2) Similarity prediction. The protein language model generates deep sequence embeddings for query and target proteins. Subsequently, SS-predictor predicts the similarity of all query-target pairs. (3) Search result. Finally, PLMSearch selects the similarity of the protein pairs pre-filtered by PfamClan, sorts the protein pairs based on their similarity, and outputs the search results for each query protein separately.
For the top-ranked query-target pairs, PLMAlign is used to generate local or global alignments and alignment scores. In addition, we also added parallel sequence-based Needleman-Wunsch alignment and structure-based TM-align at the end of our pipeline for users to choose.
PfamClan
PfamClan filters out protein pairs that share the same Pfam clan domain (Fig. 1a). It is worth noting that the recall rate is more important in the initial pre-filtering. PfamClan is based on a more relaxed standard of sharing the same Pfam clan domain, instead of sharing the same Pfam family domain (what PfamFamily does). This feature allows PfamClan to outperform PfamFamily in recall rate (Fig. 4) and successfully recalls high TM-score protein pairs that PfamFamily misses (Supplementary Table 11).
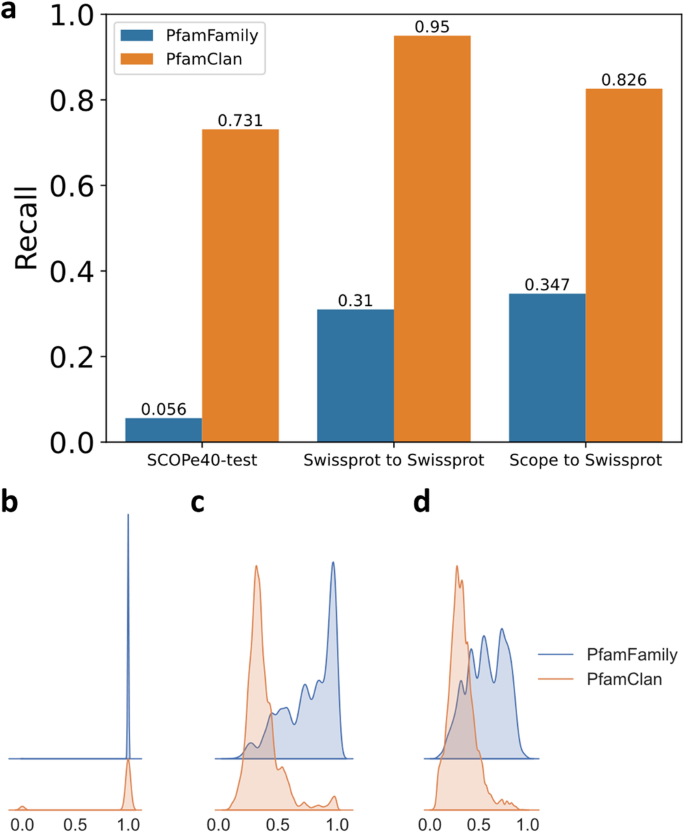
a We evaluated the pre-filtering results of PfamFamily & PfamClan on the SCOPe40-test, Swiss-Prot to Swiss-Prot, and SCOPe40-test to Swiss-Prot search tests (see “Datasets”). PfamClan achieves a higher recall rate. b Same(1) or Different(0) fold on SCOPe40-test. c–d TM-score distributions using kernel density estimation (smoothed histogram using a Gaussian kernel with the width automatically determined). c Swiss-Prot to Swiss-Prot; d SCOPe40-test to Swiss-Prot. The distribution of PfamFamily is overall to the right, because the requirements of PfamFamily are stricter than PfamClan, so the protein pair it recalls has a higher probability of being in the same fold and sharing a higher TM-score. However, this also leads to PfamFamily having a lower recall rate and missing some homologous protein pairs as shown in Supplementary Table 11. It is worth noting that the recall rate is more important in the initial pre-filtering. Source data are provided as a Source Data file.
Similarity prediction
Based on the protein language model and SS-predictor, PLMSearch performs further similarity prediction based on the pre-filtering results of PfamClan (Fig. 1b). The motivation is that the clustering results based on PfamClan show a significant long-tailed distribution (Supplementary Fig. 4). As the size of the dataset increases, the number of proteins contained in big clusters will greatly expand, further leading to a rapid increase in the number of pre-filtered protein pairs (Supplementary Table 8). The required computing resources are excessive with TM-align used for all the filtered pairs. Instead, PLMSearch employs SS-predictor to predict similarity, which increases speed and eliminates reliance on structures.
As shown in Fig. 5a, the input protein sequences are first sent to the protein language model (ESM-1b here) to generate per-residue embeddings (m*d, where m is the sequence length and d is the dimension of the vector), and the per-protein embedding (1*d) is obtained through the average pooling layer. Subsequently, SS-predictor predicts the structural similarity (TM-score) between proteins through a bilinear projection network (Fig. 5b). However, it is difficult for the predicted TM-score to directly rank protein pairs with extremely high sequence identity (often differing by only a few residues). This is because SS-predictor was trained on SCOPe40-train and CATHS40, where protein pairs share sequence identity < 0.4, so cases with extremely high sequence identity were not included. At the same time, COS similarity performs well in cases with extremely high sequence identity (see P@1 in Supplementary Fig. 3d-e) but becomes increasingly insensitive to targets after Top-10. Therefore, the final similarity predicted by SS-predictor is composed of the predicted TM-scores and the COS similarity to complement each other. We studied the reference similarity of COS similarity in SCOPe40-train (Supplementary Fig. 5a-b, Supplementary Table 12). We assemble the predicted TM-score with the top COS similarity as follows. When COS similarity > 0.995, SS-predictor similarity equals COS similarity. Otherwise, SS-predictor similarity equals the predicted TM-score multiplied by COS similarity.
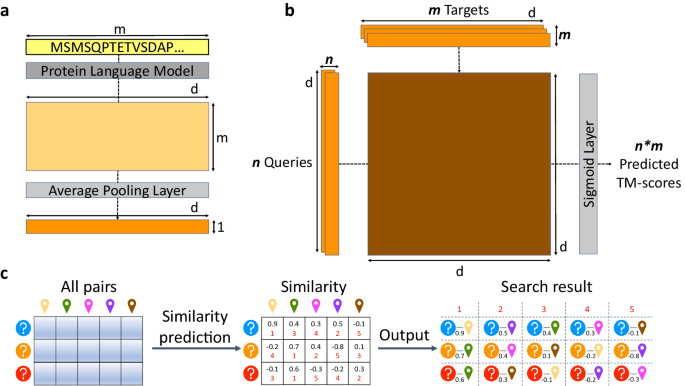
a Protein embedding generation. The protein language model converts the protein sequence composed of m residues into m*d per-residue embeddings, where d is the dimension of the embedding. Then the average pooling layer converts them into the corresponding 1*d per-protein embedding z. b SS-predictor. A bilinear projection network predicts all the TM-scores between n query embeddings z1 and m target embeddings z2 by z1*W*z2, where the matrices W are the learned parameters. c Similarity-based search methods. The similarity of all protein pairs is predicted, sorted, and then outputted as a search result. According to different similarity, three corresponding search methods are Euclidean, COS, and SS-predictor.
We also studied the reference similarity of SS-predictor similarity (Fig. 6) in SCOPe40-train. There is a clear phase transition occurring around the similarity of 0.3–0.7. Supplementary Table 12 shows that for SS-predictor, protein pairs with a similarity lower than 0.3 are usually assumed as randomly selected irrelevant protein pairs. In the ridge plot Fig. 6b, as expected, the protein pairs in the same fold and different folds are well grouped in two different similarity ranges, i.e. the protein pairs in the same fold have a higher similarity and the protein pairs in different folds have a lower one. However, since similarity and SCOP fold do not have a one-to-one correspondence, there is a small overlap. Furthermore, unlike Foldseek, which focuses on local similarity, the similarity of SS-predictor, like TM-score, focuses on global similarity (Supplementary Table 13).
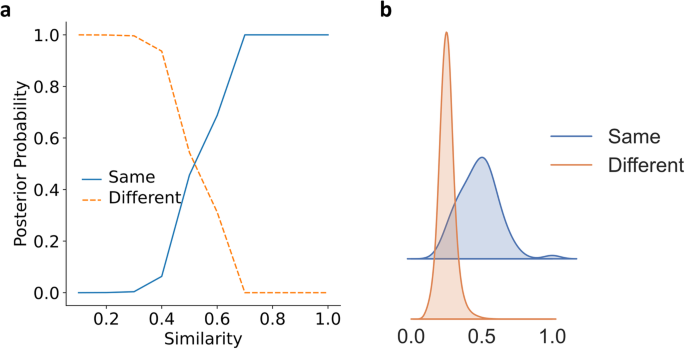
a The posterior probability of proteins with a given similarity being in the same fold or different folds in SCOPe40-train. b Similarity distribution of the same and different folds protein pairs using kernel density estimation (smoothed histogram using a Gaussian kernel with the width automatically determined). The similarity of protein pairs belonging to the same protein pair is significantly higher than that of protein pairs belonging to different folds. The posterior probability corresponding to the similarity is shown in Supplementary Table 12. See “Reference similarity” Supplement Section for more details.
PLMAlign
For the retrieved protein pairs, PLMAlign takes per-residue embeddings as input to obtain specific alignments and alignment scores (Fig. 1d). PLMAlign subsequently uses alignment scores to rerank, which improves the ranking results further. Specifically, inspired by pLM-BLAST48, PLMAlign calculates the substitution matrix by dot producting the per-residue embeddings of the query-target protein pair. The substitution matrix is then used in the SW/NW algorithm to perform local/global alignment, and the algorithm is accelerated through the linear gap penalty. Compared with traditional SW/NW using a fixed substitution matrix, the substitution matrix calculated by PLMAlign uses protein embedding generated from the sequence context, thus containing deep evolutionary information. Compared with pLM-BLAST, by using the dot product and the linear gap penalty, PLMAlign can better align remote homology pairs while reducing the algorithm complexity to O(mn) to ensure high efficiency (Supplementary Table 14). Therefore, PLMAlign performs better on remote homology alignment (using “Malisam and Malidup” datasets as benchmarks, see Supplementary Fig. 6, Supplementary Table 15). Also, see “Remote homology alignment” Supplementary Section for detailed settings of PLMAlign and the evaluation of alignment results. The reference score of PLMAlign is provided in Supplementary Fig. 5c, d and Supplementary Table 12. PLMAlign in the main text uses global alignment to generate alignment scores.
Datasets
The data volumes and uses of each dataset are summarized in Supplementary Table 16.
SCOPe40
The SCOPe40 dataset consists of single domains with real structures. Clustering of SCOPe 2.0155,56 at 0.4 sequence identity yielded 11,211 non-redundant protein domain structures (SCOPe40). As done in Foldseek, domains from SCOPe40 were split 8:2 by fold into SCOPe40-train and SCOPe40-test, and then domains with a single chain were reserved. It is worth mentioning that each domain in SCOPe40-test belongs to a different fold from all domains in SCOPe40-train, so the difference between training and testing data is much larger than that of pure random division. We also studied the max sequence identity of each protein in SCOPe40-test relative to the training dataset (Supplementary Fig. 7). From the figure, we can draw a similar conclusion that the sequences in SCOPe40-test and the training dataset are quite different, and most of the max sequence identity is between 0.2 and 0.3. PLMSearch performs well in SCOPe40-test (Fig. 2a–c, Supplementary Fig. 1a–c, Supplementary Table 2), implying that PLMSearch learns universal biological properties that are not easily captured by other sequence search methods13.
New protein search test
In real-world scenarios, newly discovered and unclassified proteins often play a crucial role in innovative research. To assess the effectiveness of various methods in searching these proteins, we introduced an additional search test exclusively comprising query proteins that failed to scan any Pfam domain. Specifically, we screened a total of 110 queries from the 2207 queries in the SCOPe40-test, which failed to scan any Pfam domain. In the all-versus-all search test on the SCOPe40-test dataset, we counted the MAP, P@1, and P@10 metrics with only new proteins as queries (110 proteins) and SCOPe40-test as targets (2207 proteins).
Swiss-Prot
Unlike SCOPe, the Swiss-Prot dataset consists of full-length, multi-domain proteins with predicted structures, which are closer to real-world scenarios. Because the throughput of experimentally observed structures is very low and requires a lot of human and financial resources, the number of real structures in datasets like PDB57,58,59 tends to be low. AlphaFold protein structure database (AFDB) obtains protein structure through deep learning prediction, so it contains the entire protein universe and gradually becomes the mainstream protein structure dataset. Therefore, in this set of tests, we used Swiss-Prot with predicted structures from AFDB as the target dataset.
Specifically, we downloaded the protein sequence from UniProt9 and the predicted structure from the AlphaFold Protein Structure Database31. A total of 542,317 proteins with both sequences and predicted structures were obtained. For these proteins, we dropped low-quality proteins with an avg. pLDDT lower than 70, and left 498,659 proteins. In order to avoid possible data leakage issues, like SCOPe40, we used 0.4 sequence identity as the threshold to filter homologs in Swiss-Prot from the training dataset. Specifically, we used the previously screened 498,659 proteins as query proteins and SCOPe40-train as the target dataset. We first pre-filtered potential homologous protein pairs with MMseqs2 and calculated the sequence identity between all these pairs. The query protein from Swiss-Prot will be discarded if the sequence identity between the query protein and any target protein is greater than or equal to 0.4. Finally, 68,519 proteins were deleted via homology filtering, leaving 430,140 proteins in Swiss-Prot, which we employed in our experiments. We also studied the max sequence identity of each protein in Swiss-Prot relative to the training dataset (Supplementary Fig. 7) and found that the vast majority of them were below 0.3.
Subsequently, we randomly selected 50 queries from Swiss-Prot and 50 queries from SCOPe40-test as query proteins (a total of 100 query proteins) and searched for 430,140 target proteins in Swiss-Prot. Therefore, a total of 43,014,000 query-target pairs were tested. The search test for 50 query proteins from Swiss-Prot and SCOPe40-test are called “Swiss-Prot to Swiss-Prot” and “SCOPe40-test to Swiss-Prot”, respectively.
CATHS40
The SCOPe40-train dataset includes 8953 proteins and TM-scores for all protein pairs were calculated for training. As the majority of these pairs had TM-scores below 0.5, only 504,553 pairs (among 80,156,209 in total) had a TM-score above 0.5 for model training. To enhance the model’s generality, we supplemented it with high-quality protein pairs extracted from the curated CATH domain dataset44,47. We began with the CATHS40 non-redundant dataset of protein domains, which exhibits no more than 0.4 sequence similarity. Domains exceeding 300 residues were filtered out, leaving 27,270 domains. To prevent potential data leakage issues, akin to SCOPe40, we applied a 0.4 sequence identity threshold to filter homologs in CATHS40 from the testing dataset (SCOPe40-test and Swiss-Prot). Finally, 21,474 proteins in CATHS40 were left for training, and the max sequence identity of the test dataset to the new training dataset is still less than 0.4 (Supplementary Fig. 7). We then undersampled CATHS40 domain pairs from different folds to acquire a substantial amount of training pairs with TM-scores above 0.5. Specifically, we sampled the TM-scores of 28,440,312 protein pairs for training, of which 7,813,946 pairs had a TM-score above 0.5 (Supplementary Table 16).
Target datasets on the web server
We currently have the following four target datasets on the web server for users to search: (1) Swiss-Port (568K proteins)9, the original dataset without filtering; (2) PDB (680K proteins)57,58,59; (3) UniRef50 (53.6M proteins)9. UniRef50 is built by clustering UniRef90 seed sequences that have at least 50% sequence identity to and 80% overlap with the longest sequence in the cluster; (4) Self (the query dataset itself).
Malisam and Malidup
We employed two gold-standard benchmark datasets, Malisam60 and Malidup61, to establish a robust reference for alignment. These sets rigorously select structural alignments, emphasizing difficult-to-detect, low-sequence-identity remote homology. Malidup contains pairwise structure alignments, specifically targeting homologous domains within the same chain, thereby exemplifying structurally similar remote homologs. Malisam contains pairs of analogous motifs.
Metrics
We evaluated different homologous protein search methods using the following four metrics: AUROC, AUPR, MAP, and P@K.
-
AUROC24: The mean sensitivity over all queries, where the sensitivity is the fraction of TPs in the sorted list up to the first FP, all excluding self-hits.
-
AUPR24: Area under the precision-recall curve.
-
MAP62: Mean average precision (MAP) for a set of queries is the mean of the average precision scores for each query.
$$MAP=frac{mathop{sum }nolimits_{q=1}^{Q}AveP(q)}{Q}$$
(1)
where Q is the number of queries.
-
P@K20: For homologous protein search, as many queries have thousands of relevant targets, and few users will be interested in getting all of them. Precision at k (P@k) is then a useful metric (e.g., P@10 corresponds to the number of relevant results among the top 10 retrieved targets). P@K here is the mean value for each query.
On SCOPe40-test, we performed an all-versus-all search test, which means both the query and the target dataset were SCOPe40-test. To make a more objective comparison, the settings used in the all-versus-all search test are exactly the same as those used in Foldseek24. Specifically, for family-level, superfamily-level, and fold-level recognition, TPs were defined as the same family, same superfamily but different family, and same fold but different superfamily, respectively. Hits from different folds are FPs. After sorting the search result of each query according to similarity (described in Supplementary Table 17), we calculated the sensitivity as the fraction of TPs in the sorted list up to the first FP to better reflect the requirements for low false discovery rates in automatic searches. We then took the mean sensitivity over all queries as AUROC. Additionally, we plotted weighted precision-recall curves (precision = TP/(TP+FP) and recall = TP/(TP+FN)). All counts (TP, FP, FN) were weighted by the reciprocal of their family, superfamily, or fold size. In this way, families, superfamilies, and folds contribute linearly with their size instead of quadratically14. MAP and P@K were calculated according to the TPs and FPs defined by fold-level.
On search tests against Swiss-Prot, without the human manual classification on SCOPe as the gold standard, proteins require a reference annotation method. Therefore, TPs were defined as pairs with a TM-score > 0.5, otherwise FPs. MAP and P@K are then calculated accordingly.
Baselines
Previously proposed methods
(1) Sequence search: MMseqs210, BLASTp11, HHblits16,17, EAT45, and pLM-BLAST48. (2) Structure search—structural alphabet: 3D-BLAST-SW22, CLE-SW23, Foldseek, and Foldseek-TM24. (3) Structure search—structural alignment63: CE25, Dali26, and TM-align27,28. For the specific introduction and settings of these proposed methods, see “Baseline details” Supplementary Section.
Similarity-based search methods
These methods predict and sort the similarity between all query-target pairs (Fig. 5c). Different search methods are distinguished according to the way they predict similarity.
-
Euclidean: Use the reciprocal of the Euclidean distance between embeddings.
$$similarity(p,q)=frac{1}{sqrt{mathop{sum }nolimits_{i=1}^{n}{left({p}_{i}-{q}_{i}right)}^{2}}+1}$$
(2)
-
COS: Use the COS distance between embeddings. ϵ is a small value to avoid division by zero.
$$similarity(p,q)=frac{pcdot q}{max left({leftVert prightVert }_{2}cdot {leftVert qrightVert }_{2},epsilon right)}$$
(3)
-
SS-predictor: Combine the predicted TM-score with the top COS similarity. Unlike PLMSearch, which predicts pre-filtered pairs from PfamClan, SS-predictor extends its prediction to all protein pairs.
Experiment settings
Pfam result generation
We obtained the Pfam family domains of proteins by PfamScan (version 1.6)54 and Pfam dataset (Pfam36.0, 2023-09-12)42. For PfamClan, we query the comparison table Pfam-A.clans.tsv and replace the family domain with the clan domain it belongs to. For the family domain that has no corresponding clan domain, we treat it as a clan domain itself.
Protein language model
ESMs are a set of protein language models that have been widely used in recent years. We used ESM-1b (650M parameters)35, a SOTA general-purpose protein language model, to efficiently generate per-protein embeddings for PLMSearch.
For PLMAlign, the more extensive ProtT5-XL-UniRef50 (3B parameters) is used to generate per-residue embeddings. We conducted a detailed evaluation and analysis of the results from ESM-1b and ProtT5-XL-UniRef50 embeddings, as elaborated in “Remote homology alignment” Supplementary Section (Supplementary Fig. 8, Supplementary Table 15). Based on the analysis, for PLMAlign, ProtT5-XL-UniRef50 was selected.
SS-predictor training
We used the deep learning framework PyTorch (version 1.7.1), ADAM optimizer, with MSE as the loss function to train SS-predictor. The batch size was 100, and the learning rate was 1e-6 on 200 epochs. The training ground truth was the TM-score calculated by TM-align. The datasets used for training (Supplementary Table 16) include (1) All protein pairs from SCOPe40-train (8953 proteins; 80,156,209 query-target pairs). (2) Undersampled protein pairs from CATHS40 (21,474 proteins; 28,440,312 query-target pairs). The parameters in ESM-1b were frozen and only the parameters in the bilinear projection network were trained.
Experimental environment
We conducted the experiments on a server with a 56-core Intel(R) Xeon(R) CPU E5-2680 v4 @ 2.40 GHz and 256 GB RAM. The environment of our publicly available web server is 64 * Intel(R) Xeon(R) CPU E5-2682 v4 @ 2.50 GHz and 512 GB RAM.
Statistics and reproducibility
The experiments were not randomized.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-024-46808-5