Ethical approval
The Institutional Animal Care and Use Committee (IACUC, 22-422) and NIH Guide for the Care and Use of Laboratory Animals were adhered to in all animal research. All studies and protocols were approved by IACUC of North Carolina State University (protocol: 19-811-B).
Nanovial fabrication
Polyethylene glycol (PEG) biotinylated nanovials with 50 μm diameters were fabricated using a three-inlet flow-focusing microfluidic droplet generator, sterilized and stored at 4 °C in Washing Buffer consisting of Dulbecco’s Phosphate Buffered Saline (Thermo Fisher) with 0.05% Pluronic F-127 (Sigma), 1% 1X antibiotic-antimycotic (Thermo Fisher), and 0.5% bovine serum albumin (Sigma) as previously reported16,17,18,42. In brief, PEG pre-polymer, gelatin, and oil phases were infused into a droplet generator microfluidic device and polymerized under UV light through a DAPI filter set on an inverted microscope. Polymerized nanovials were collected in a conical tube and washed to remove unpolymerized phases. A total diameter and inner cavity diameter were calculated using a MATLAB script as reported in our previous study. The inner cavity size for 50 μm nanovials was 30 μm ± 2 μm on average. As the average size of MSCs is heterogenous (15–30 μm in diameter), the largest diameter of MSCs (30 μm) was considered when choosing the nanovial size (50 μm)16,17,18,42.
Nanovial functionalization
Streptavidin conjugation to the biotinylated cavity of nanovials
50 μm sterile nanovials were diluted in Washing Buffer five times the volume of the nanovials (i.e., 100 μL of nanovial volume was resuspended in 400 μL of Washing Buffer). A diluted nanovial suspension was incubated with an equal volume of 200 μg/mL of streptavidin (Thermo Fisher) for 30 min at room temperature on a tube rotator. Excess streptavidin was washed out three times by pelleting nanovials at 2000 × g for 30 s on a Galaxy MiniStar centrifuge (VWR), removing supernatant, and adding 1 mL of fresh Washing Buffer.
Anti-CD63 EV capture antibody labeled nanovials
50 μm streptavidin-coated nanovials were reconstituted at a five-time dilution in a Washing Buffer containing 270 nM (40 μg/mL) of biotinylated anti-CD63 antibody (Biolegend, 353018). Nanovials were incubated with antibodies for 30 min at room temperature on a rotator and washed three times as described above. Nanovials were resuspended at a five times dilution in a Washing Buffer or culture medium prior to each experiment.
Cell culture
Immortalized mesenchymal stem cells (iMSC)
hTERT immortalized adipose-derived MSCs (ATCC, ASC52telo) were cultured based on the manufacturer’s protocol using MSC basal media (ATCC, PCS500030) and growth kit (PCS500040). For every secretion assay in this study, cells at passage 18 were used to maintain consistency.
C57BL/6 Mouse Bone Marrow Mesenchymal Stem Cells. Mouse mesenchymal stem cells (cell biologics, C57-6043) were maintained in Iscove’s modified Dulbecco’s medium (IMDM) (Invitrogen) containing 10% fetal bovine serum (FBS). Cells at passage 8 were used in the in vivo study.
HL-1 cells (Sigma, SCC065) were maintained in Claycomb Medium (Sigma) containing 10% fetal bovine serum (FBS). Cells at passage 6 were used in the in vitro study.
Nanovial secretion assay general procedure
Cell loading onto nanovials
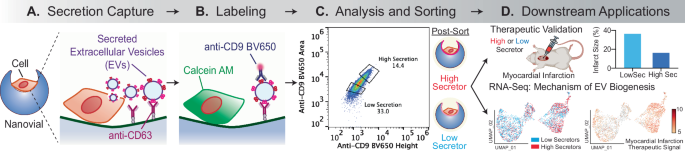
Each well of a 24-well plate was filled with 1 mL of media and 50 μL of 5× reconstituted functionalized nanovials (10 μL of nanovial volume = 100,000 total nanovials) was added in each well using a standard micropipette. Cells were seeded in each well and extra culture medium was added to make a total volume of 1.5 mL. Each well was mixed by simply pipetting 5 times with a 1000 μL pipette set to 1000 μL. The well plate was transferred to an incubator to allow cell binding; the volume in each well was pipetted up and down again 5 times with a 200 μL pipette set to 200 μL at 30-min intervals. Repeated resuspension of nanovials and cells ensured unbound cells that initially did not settle into nanovial cavities in the first loading step could bind to empty nanovials when remixed. Cells that have fallen into a cavity are given 30 min to form initial integrin-based adhesions to the gelatin in the nanovial. After one hour, nanovials were strained using a 20 μm cell strainer to remove any unbound cells and recovered. During this step, any unbound cells were washed through the strainer and only the nanovials (with or without cells loaded) were recovered into a 12-well plate with 2 mL of media by inverting the strainer and flushing with media.
Secretion accumulation and secondary antibody staining on nanovials
After cell loading, cells on nanovials were incubated for 24 h in the incubator to accumulate secretion. Each sample was recovered in a conical tube with 5 mL wash buffer and centrifuged for 5 min at 200 × g. The supernatant was removed and nanovials were reconstituted at a ten-fold dilution in a Washing Buffer containing detection antibody and/or cell viability dye (calcein AM) to label secreted EVs and viability of cells. For each 100,000 nanovials (~10 μL nanovial volume), 5 μL of anti-CD9 BV650 antibody with 85 μL of 0.3 μM Calcein AM solution, unless otherwise stated. Nanovials were incubated with the detection antibody at 37 °C for 30 min, protected from light. After washing nanovials with 5 mL of Washing Buffer, nanovials were resuspended at a 40-fold dilution in the Washing Buffer and transferred to a flow tube.
Flow cytometer analysis and sorting
All flow cytometry sorting was performed using the SONY SH800 cell sorter equipped with a 130-micron sorting chip (SONY Biotechnology). The cytometer was configured with violet (405 nm), blue (488 nm), green (561 nm), and red (640 nm) lasers with 450/50 nm, 525/50 nm, 600/60 nm, 665/30 nm, 720/60 nm and 785/60 nm filters. In each analysis, samples were compensated using negative (blank nanovials) and positive controls (purified EV conjugated nanovials labeled with each fluorescent detection antibody or cells stained with calcein AM). Nanovial samples were diluted to approximately 623 nanovial/μL in Washing Buffer for analysis and sorting. Drop delay was configured using standard calibration workflows and single-cell sorting mode was used for all sorting as was previously determined to achieve the highest purity and recovery43. A sample pressure of 4 was set for all analysis and cell sorting processes. The following gating strategy was used to identify cells on nanovials with strong secretion signal: (1) nanovial population based on high forward scatter height and side scatter area, (2) calcein AM positive population, (3) CD9 EV secretion signal positive population based on fluorescence peak area and height.
For analyzing samples using the ImageStream imaging-based flow cytometer, a 10 μL nanovial sample was resuspended in 200 μL of Washing Buffer and analyzed based on the manufacturer’s protocol. Images were analyzed by IDEAS software distributed by Luminex. Analysis with the ImageStream was repeated two times with three biological replicates for each experiment.
EV secretion phenotype analysis using ExoView
Human iMSCs were seeded in a T-75 flask at a seeding density of 0.2 million cells per flask with stem cell basal media with 2% FBS. After 24 h of initial seeding, media was changed to OPTI-MEM I reduced serum media without phenol red (Fisher Scientific, 11-058-021). After 48 h, conditioned media was centrifuged at 2500 × g for 10 min at 4 °C and the supernatant was filtered using a 0.2 μm syringe filter (VWR, 28143-310). The sample placed in ultracentrifuge tubes (Fisher Scientific, NC9732446) was placed into a pre-cooled 50 Ti fixed angle titanium rotor and centrifuged in Optima XPN-100 Ultracentrifuge at 120,000 × g for 120 min at 4 °C. Media was removed and the pellet was resuspended in 100 μL cold PBS. The sample was analyzed by the ExoView R100 for CD63, CD9, and CD81 markers based on the manufacturer’s protocol. The experiment comprised three biological replicates. For profiling mouse tetraspanin markers, we analyzed EVs concentrated from mouse bone-marrow-derived MSCs using the ExoView mouse tetraspanin kit (CD81 and CD9 capture and CD81, CD63, CD9 as fluorescent counter stains) at the Extracellular Vesicle Core, Children’s Hospital Los Angeles.
Dynamic range of CD63+CD9+ EVs on nanovials
Nanovials were labeled with biotinylated anti-CD63 antibody using the modification steps mentioned above and incubated with EVs isolated from ultracentrifugation at 0, 30, 60, or 120 μg/mL for 2 h at 37 °C. Excess EVs were removed by washing nanovials three times with a Washing Buffer. Nanovials were pelleted at the last wash step and incubated with anti-CD9 as described in the secondary antibody staining procedure above. Following washing three times, nanovials were reconstituted at a 50 times dilution in the Washing Buffer and transferred to a flow tube. The fluorescent signal on nanovials was analyzed using a cell sorter with sensors and imaged using a fluorescence microscope. This experiment was repeated two times to confirm the reproducibility of EV capture on nanovials.
Single-MSC loading and statistics
50 μm nanovials labeled with anti-CD63 antibodies were prepared using the procedures described above. To test cell concentration-dependent loading of nanovials, 0.1 × 106 (1 cell per nanovial) and 0.2 × 106 (2 cells per nanovial) of cell tracker deep green stained cells were each seeded onto 100,000 nanovials in a 24-well plate, and recovered as described above. Samples resuspended at 40-fold dilution in Washing buffer were analyzed using a SONY sorter for loading efficiency. The population in each gate (single-cell-loaded nanovial, multiple-cell-loaded nanovial, nanovial aggregate) was sorted into 96-well plate containing Washing buffer and imaged using a fluorescence microscope.
Analysis and sorting of single cells based on CD63+CD9+ EV secretion level
0.1 million MSCs were loaded onto anti-CD63 labeled nanovials and secretion was accumulated for 24 h. The same number of cells were also loaded onto unlabeled nanovials (no capture antibody) as a negative control. After 24 h secreted EVs were labeled with fluorescent anti-CD9 antibodies and calcein AM viability dye. After resuspending nanovials at 40-fold dilution in Washing Buffer, a small fraction of the sample was transferred to a 96-well plate to be imaged using a fluorescence microscope prior to sorting. Samples were analyzed using a cell sorter based on a combination of fluorescence area and height signals. To sort live single cells based on secretion signal, nanovials with calcein AM staining were first gated and high, medium, or low secretors and sorted by thresholding above the negative control sample on CD9 fluorescence area and height signals. Sorted samples were imaged with a fluorescence microscope to validate the enrichment of nanovials based on the amount of secreted EVs captured on the nanovials. This experiment was repeated three times to confirm the reproducibility and isolation of corresponding high and low EV secretors.
Expansion of cells sorted based on EV secretion level
In each well of a 96-well plate filled with 150 μL stem cell basal media, a single nanovial containing a single cell with a high EV secretion signal was sorted (n = 96). In another two 96-well plates, for each well one nanovial containing single cells with high or medium secretion signal was also sorted. For sorting groups of 3000 cells with the same secretion phenotype, 3000 single-cell-loaded nanovials were sorted based on high, medium, or low secretion gates into each well of a new 96-well plate. Since low secretors proliferated much slower than medium and high secretors, cells were expanded till Day 13 and re-seeded into the T-75 flask at 0.2 million cells per flask. Similarly, the single-cell colony was expanded till Day 22 and re-seeded into T-75 flask at the same seeding density. 24 h after cells were seeded, the media was changed to OPTI-MEM reduced serum media. After 48 h (Day 25 for single-cell colony and Day 16 for bulk-sorted population), conditioned media was collected, and the final number of cells was counted. EVs were collected from conditioned media via ultracentrifugation and quantified by ZetaView analysis at the University of North Carolina Nanomedicines Characterization Core Facility. For calculating EV production efficiency, the total EV count was divided by the total number of cells in each sample.
Single-cell transcriptomic analysis
The standard protocol for 10X Chromium single-cell 3′ GEX (Chromium Next GEM Single-Cell 3′ Kit v3.1 from 10X Genomics, Catalog number: PN-1000268) was followed unless otherwise noted. Sorted samples reconstituted at 18 μL were loaded into the 10X Chromium Next GEM Chip for partitioning each nanovial or cell into droplets allowing the PCR amplification and enrichment of gene expression for individual cell barcoded cDNA. Single-cell libraries were constructed using the manufacturer-recommended protocol by the UCLA Technology Center for Genomics & Bioinformatics. Libraries were then sequenced on Novaseq SP (235–400 M/lane) (Illumina). The Cell Ranger pipeline (Cell Ranger Count v7.0.0 with reference (Human (GrCh38) 2020-A) from 10X cloud analysis system) was used for sample de-multiplexing. 10X Loupe browser was used for further analysis and generation of UMAP and violin plots. For EV biogenesis (GO: 0140112) and stem cell proliferation signatures (GO: 2000648), a gene list was downloaded from the Jackson Laboratory. For tissue regeneration and vascular regenerative signatures, a gene list was retrieved from previous studies25,28.
In vitro differentiation of iMSC and iMSC loaded into nanovials
Cell sorting and expansion
A human mesenchymal stem cell functional identification kit (R&D systems, SC006) was utilized for functional characterization and comparisons of iMSCs and iMSCs loaded onto nanovials. First, cultured passage 16 iMSCs that had reached 80% confluency were enzymatically harvested with TrypLE (Gibco, 1260413). Half the cells were re-plated for continued culture in a flask while the remaining cells were loaded into nanovials according to the procedures described earlier. Live cells on nanovials were stained with Calcein AM (Invitrogen, C3099) and sorted (Sony Biotechnology, SH800S) into a 24-well plate pre-filled with MSC culture media (8000 events/well). The cells on nanovials were grown in the well plate for a few days until they reached 90% confluency (culture media was changed every 3 days). Upon reaching the desired confluency, the cells were washed once with PBS and detached with TrypLE. The mixture of detached cells and nanovials was then passed through a 37 µm strainer (STEMCELL Technologies, 27215). The nanovials were captured by the strainer and the cells were collected in a 15 ml conical tube, centrifuged, resuspended in MSC media, and counted. In addition, the original cells which were cultured in a flask and grown to confluency (cells that were not loaded into nanovials) were also sorted and expanded in the same manner.
Induction of adipogenic differentiation and FABP4 staining
Resuspended cells from nanovials as well as the original cells were seeded into separate wells at a density of 3.7 × 104 cells/well. The cells were washed into α MEM Basal Media (R&D Systems, CCM007) containing 10% FBS and 1% anti-anti prior to seeding. The plate was incubated for a day in a 37 °C and 5% CO2 incubator. Once the cells reached 100% confluency, the media was replaced with adipogenic differentiation media (α MEM Basal Media containing adipogenic supplement) to induce differentiation. Media in control wells was replaced with fresh α MEM Basal Media only. The media replacement was repeated once every 3 days with freshly prepared media. The culture was continued until day 21 at which point the media was aspirated, cells were washed twice with PBS, and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were then washed twice in 1% BSA solution in PBS, and permeabilized and blocked in 0.5 ml of 0.3% Triton X-100, 1% BSA, and 10% normal donkey serum in PBS at room temperature for 45 min. Next, the cells were incubated in 10 µg/mL anti-mFABP4 primary antibody solution overnight at 4 °C. Negative staining wells were also prepared without the primary antibody solution. The next day, cells were washed three times with 1% BSA in PBS and stained with 1:200 diluted NL557-conjugated donkey anti-goat secondary antibody (R&D systems, NL001) in 1% BSA in PBS for 60 min. The cells were then washed three times with 1% BSA in PBS and stained with 1 µg/mL DAPI solution (Thermo Fisher Scientific, 62248) for 4 min. The samples were washed 3 additional times with 1% BSA and placed in PBS for microscopic imaging.
Induction of osteogenic differentiation and anti-hOsteocalcin staining
Resuspended cells from nanovials as well as the original cells were seeded into separate wells at a density of 7.4 × 103 cells/well. The cells were washed into α MEM Basal Media (R&D Systems, CCM007) containing 10% FBS and 1% anti-anti prior to seeding. The plate was incubated for a day in a 37 °C and 5% CO2 incubator. Once the cells reached 70% confluency, the media was replaced with osteogenic differentiation media (α MEM Basal Media containing osteogenic supplement) to induce differentiation. Media in control wells was replaced with fresh α MEM Basal Media only. The media replacement was repeated once every 3 days with freshly prepared media. The culture was continued until day 21 at which point the media was aspirated, cells were washed twice with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were then washed three times with 1% BSA solution in PBS, and permeabilized and blocked in 0.5 ml of 0.3% Triton X-100, 1% BSA, and 10% normal donkey serum in PBS at room temperature for 45 min. Next, the cells were incubated in 10 µg/mL anti-hOstetocalcin primary antibody solution overnight at 4 °C. Negative staining wells were also prepared without the primary antibody solution. The next day, cells were washed three times with 1% BSA in PBS and stained with 1:200 diluted NL557-conjugated donkey anti-mouse secondary antibody (R&D systems, NL007) in 1% BSA in PBS for 60 min. The cells were then washed three times with 1% BSA in PBS and stained with 1 µg/mL DAPI solution (Thermo Fisher Scientific, 62248) for 4 min. The samples were washed 3 additional times with 1% BSA and placed in PBS for microscopic imaging.
Induction of chondrogenic differentiation and anti-hAggrecan staining
Resuspended cells from nanovials as well as the original cells were seeded into separate 15 ml conical tubes at a count of 2.5 × 105 cells/tube. The cells were centrifuged at 200 × g for 5 min at room temperature. The media was removed and replaced with D-MEM/F-12 Basal Media (Gibco, 11320033) containing 1% ITS supplement and 1% anti-anti and centrifuged at 200 × g for 5 min. This procedure was repeated once more. After the last wash, the cells were placed in chondrogenic differentiation media (D-MEM/F-12 Basal Media containing chondrogenic supplement) to induce chondrogenic differentiation. Media in control tubes was replaced with fresh D-MEM/F-12 Basal Media only. The tubes were centrifuged once more at 200 × g for 5 min at room temperature and placed in a 37 °C and 5% CO2 incubator. The media replacement was repeated once every 3 days with freshly prepared media without disturbing the pellet. The culture was continued until day 21 at which point the media was aspirated, each pellet was washed twice with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. The pellet was then washed two times with PBS, molded into a cryosectioning mold with Tissue-Tek OCT compound (Sakura, 4583), flash frozen with liquid nitrogen vapor, and stored at −80 °C until cryosectioning was performed. 5 µm-thick sections were obtained using cryostat microtome (Lecia CM1950) and kept frozen until the staining procedure. To stain the pellet sections, the pellet sections were permeabilized and blocked in 0.5 ml of 0.3% Triton X-100, 1% BSA, and 10% normal donkey serum in PBS at room temperature for 45 min. Next, the sections were incubated in 10 µg/mL anti-hAggrecan primary antibody solution overnight at 4 °C. Negative staining sections were also prepared without the primary antibody solution. The next day, the sections were washed three times with 1% BSA in PBS and stained with 1:200 diluted NL557-conjugated donkey anti-goat secondary antibody (R&D systems, NL001) in 1% BSA in PBS for 60 min. The sections were then washed three times with 1% BSA in PBS and stained with 1 µg/mL DAPI solution (Thermo Fisher Scientific, 62248) for 4 min. The samples were washed 3 additional times with 1% BSA, followed by washing once with distilled water. Excess water was removed, and the sections were covered with a drop of mounting medium and a glass coverslip for microscopic imaging.
Scanning electron microscopy of nanovials
After FACS sorting, nanovials containing both high and low EV-secreting MSCs were subjected to fixation using 4% paraformaldehyde (PFA) (Thermo Fisher, J61899) for a duration of 20 min. Following fixation, the nanovials were thoroughly washed with DI water and subsequently dried at 37 °C on a silicon wafer. Post-drying, a gold coating was applied to the nanovials, enabling the characterization of cell-loaded nanovial morphology through Scanning Electron Microscopy (SEM) (JCM-7000, JEOL).
In vitro ischemic injury model
To establish the ischemic injury in vitro, HL-1 cells were incubated with 500 μM H2O2 in a complete medium for 2 h. After that, the H2O2-containing medium was replaced by a complete medium with EVs isolated from the same number of MSCs with different secretion abilities. For 24 h treatment, the cell apoptosis was evaluated using TUNEL staining44.
IPC injection of MSCs in mice model of MI
The Institutional Animal Care and Use Committee (IACUC, 22-422) and NIH Guide for the Care and Use of Laboratory Animals were adhered to in all animal research. All mice are housed under a 12-h light/12-h dark cycle at a constant temperature of 25 °C, with humidity maintained between 40–60%. To create a mouse MI model, C57BL/6 (Charles River, C57BL/6NCrl, 8 weeks) mice were anesthetized through intraperitoneal injection of K‐X cocktail (100 mg/kg ketamine and 10 mg/kg xylazine), while a small animal ventilator (SAR‐1000 Small Animal Ventilator, CWE, Inc.) provided artificial ventilation as life support. The LAD coronary artery was ligated permanently with a 6-0 suture under sterile conditions to induce ischemia. Successful induction of Infarction was confirmed by the pale color observed in the apex area. To avoid sex bias, each experimental group comprised 4 male mice and 3 female mice. For the control group, not all mice survived in the first 7 days following the procedure, so this group comprised 12 mice initially. The 5 mice that did not survive by day 7 were excluded from echocardiography and histology studies as they did not make it to the subsequent time points (day 14 and day 28). Immediately after MI induction, 0.2 million cells with 10 µL 10 mg/mL 4% hyaluronic acid (HA) hydrogel were injected into the pericardial cavity of each mouse heart. The chest was then closed, and the animal was permitted to recover.
Mouse echocardiology
Echocardiography was carried out at various time points, including baseline (prior to MI surgery), and 2, 14, and 28 days after the MI procedure. To conduct transthoracic echocardiography, mice were anesthetized using an isoflurane/oxygen mixture and positioned supine with the body temperature maintained at 37 °C. A 40-MHz probe from the high-frequency ultrasound system (Prospect, S-Sharp, New Taipei City, Taiwan) was used to obtain B (brightness, 2D) and M (motion) mode images. Left ventricular dimensions were measured at both diastole and systole, with five continuous cardiac cycles collected for each animal. The images were recorded and assessed blindly. The ejection fraction was calculated using the following formula: EF = (LVEDV – LVESV/LVEDV) × 100%, while the fractional shortening was determined using FS = (LVEDD – LVESD/LVEDD) × 100%.
Histology
Mice tissues were harvested and fixed using a neutral buffered 10% formalin solution (NBF, Sigma–Aldrich, St. Louis, MO, USA) overnight. Afterward, the tissues were dehydrated in a 30% sucrose solution at 4 °C for at least one night. Subsequently, the tissues were cryopreserved in optimal cutting temperature (OCT) compound and cryosectioned (CryoStats, Leica). Pathological assessments were carried out using H&E and Masson’s trichrome staining in accordance with the manufacturer’s instructions (Sigma–Aldrich, St. Louis, MO, USA).
For immunofluorescence staining, cryosections of the tissues were first permeabilized and blocked using a protein block solution (Dako, Carpinteria, CA, USA) containing 0.1% saponin (Sigma–Aldrich, St. Louis, MO, USA). The primary antibodies were then added and incubated overnight at 4 °C, including rabbit anti-Caspase-3 (1:100; ab184787, Abcam, Cambridge, UK), rabbit anti-Ki67 (1:100; ab15580, Abcam), anti-CD31 antibody (1:100; EPR17259, Abcam), and mouse Anti-Cardiac Troponin T antibody (1:100; ab8295, Abcam) to target proteins of interest. Fluorophore-conjugated secondary antibodies were then added for fluorescent imaging. All the tissue slides were mounted by ProLong gold antifade mountant with DAPI (Thermo Fisher, US) before being imaged using an epifluorescent microscope from Olympus. This experiment was performed with n = 7 biological replicates for each group.
Serum chemistry analysis
At the experimental endpoint, blood samples were collected from mice, and serum was isolated using BD Vacutainer® serum collection tubes (BD, 367814). The serum chemistry test was performed by the Department of Clinical Pathology, College of Veterinary Medicine, NC State University.
Statistics and reproducibility
All experiments were performed at least twice to ensure reproducibility. Analysis and sorting of iMSCs based on EV secretion was repeated three times independently to confirm consistent outputs. We have determined the sample size for the in vivo protocol based on a power calculation (power = 0.9, effect size = 0.9, significance level = 0.05). n = 7 per group was large enough to compare three experimental groups. One-way analysis of variance (ANOVA), followed by Tukey’s honestly significant difference (HSD) post hoc test was performed with p < 0.05, unless otherwise stated. Single-cell RNA sequencing data was analyzed using 10X Genomics Cell Ranger v7.0.0 on 10X Cloud online software, and plots were generated using the 10X Genomics Loupe browser. No statistical method was used to predetermine sample size and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-024-49123-1