Animals
All animal experiments were carried out in accordance with institutional animal care protocols and were authorized by Anhui Medical University Ethical Committee (No. LLSC20160052) starting from March 1st, 2018. Adult female Sprague–Dawley (SD) rats (7–8 weeks old and weighing 200–250 g) were supplied from Experimental Animal Center of Anhui Medical University (No. SCXK (Wan) 2017-001). Animals were housed in a room with a 12-h light/dark cycle at 23–25 °C. All animals had free access to water and food. They were randomized to subsequent experiments as random.
Experimental design
For this study, BMSCs overexpressing hBcl2 (referred to as the hBcl2 group), BMSCs overexpressing hBcl2 with an endoplasmic reticulum-anchored segment (hBcl2-cb) (referred to as the cb group), and a negative control group (referred to as the NC group) were constructed by lentivirus. The expression of hBcl2 and its effect on BMSCs’ phagocytosis of myelin debris and post-transplantation survival were verified using immunocytochemistry staining. Furthermore, the relationship between the anti-apoptotic ability of BMSCs overexpressing hBcl2 and the lysosomal pathway was explored. Finally, the effect of BMSCs overexpressing hBcl2 on functional recovery in rats with spinal cord injury was observed.
Isolation and characterization of BMSCs
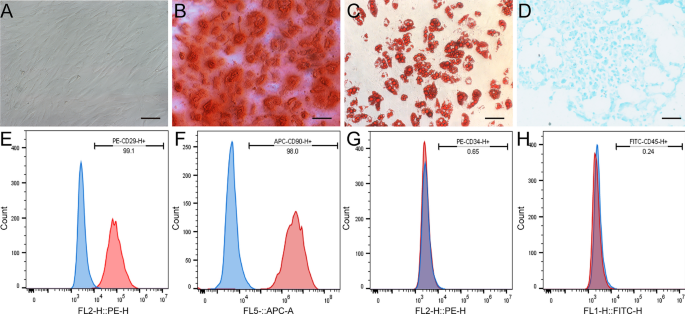
The isolation and phenotypic characterization of rat BMSCs were carried out according to previously established protocols16. Five adult female SD rats were used for BMSCs isolation. BMSCs suspensions were extracted from the distal femur and proximal tibia of the rats by repeatedly flushing the medullary cavity with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% Penicillin–Streptomycin (Gibco, USA). The total cell suspension was then cultured at 37 °C with 5% CO2 in a humidified incubator. The culture media was changed every 3 days to remove the non-adherent cells. Cells from passages 3 to 6 were collected for subsequent experiments.
The expression of CD29 (ab218273-PE, abcam, United Kingdom), CD90 (ab25322-APC, abcam, United Kingdom), CD34 (ab134207-PE, abcam, United Kingdom), and CD45 (ab27287-FITC, abcam, United Kingdom) in BMSCs was detected by flow cytometry (FACSCanto™, USA) for BMSCs phenotype characterization. Chondrogenic, osteogenic, and adipogenic differentiation were performed to determine the stem cell’s ability of BMSCs by using a three-line differentiation kit (Cyagen, China) following the manufacturer’s instructions.
The following media were used to confirm the multipotential differentiation ability of rat BMSCs: (1) adipogenic differentiation medium (10% FBS, 1% Glutamine, 0.1% Insulin, 0.1% Dexamethasone, 0.1% Rosiglitazone, 1% Penicillin-Streptomycin, high-glucose DMEM); (2) chondrogenic differentiation medium (0.3% Ascorbate, 1% ITS + Supplement, 0.1% Sodium Pyruvate, 0.1% Proline, 1% TGF-3, 0.01% Dexamethasone, high-glucose DMEM); and (3) osteogenic differentiation medium (10% FBS, 1% Glutamine, 1% Glycerophosphate, 0.2% Ascorbate, 0.01% Dexamethasone, 1% Penicillin–Streptomycin, high-glucose DMEM). The media were refreshed every 3 days. Alizarin Red staining was used to detect osteocytes, Oil Red O staining for adipocytes, and Alcian Blue staining for chondrocytes.
Construction of hBcl2 overexpressing BMSCs by lentivirus
The sequence of cDNA of hBcl2 (Plasmid #17999), hBcl2-cb (Plasmid #18000) containing an endoplasmic reticulum-anchored segment cytochrome b5, were obtained from Addgene. Lentiviral vectors, including LV5-hBcl2-GFP, LV5-hBcl2-cb-GFP, and the empty lentiviral control LV5-GFP, were synthesized by GenePharma. Lentiviruses were produced by co-transfecting these lentiviral vectors individually with the packaging plasmids (pGag/Pol, pRev, pVSV-G) into 293T cells using Lipofiter (GenePharma, China).
BMSCs were seeded in sterile petri dishes (6 cm) and allowed to reach 50% confluency. Lentivirus-containing medium (multiplicity of infection = 15) combined with polybrene (5 μg/mL; GenePharma, China) was added for lentiviral infection, according to a previously published protocol16. Fresh medium was added to the culture medium after 24 h. Stably-infected BMSCs were obtained after 2 weeks of puromycin (2 μg/mL, Sigma, USA) selection. The normal uninfected BMSCs were used as negative control. The BMSCs successfully infected with the lentiviral vector LV5-hBcl2-GFP were designated as the hBcl2 overexpressing BMSCs group (hBcl2 group). BMSCs infected with the LV5-hBcl2-cb-GFP vector, were referred to as the hBcl2-cb group (cb group). BMSCs infected with the empty vector LV5-GFP were labeled as the NC group. Western blot analysis and Immunocytochemistry were applied to detect the expression levels of hBcl2 in each group.
TUNEL analysis
BMSCs from all groups were washed with PBS and then fixed using 4% paraformaldehyde (Biosharp, China). The terminal transferase-mediated dUTP fluorescein nick-end labeling (TUNEL) assay was performed using the Cell Apoptosis Detection Kit (elabscience, China) in accordance with the manufacturer’s protocol. The percentage of apoptotic cells was calculated by the proportion of TUNEL+ cells in total cells under fluorescence microscope (Zeiss, Germany). Three visual fields were randomly selected from each group for analysis.
Isolation of myelin debris
The animal experiments were authorized by Anhui Medical University Ethical Committee (No. LLSC20160052) starting from March 1st, 2018. Myelin debris was isolated from the brains of healthy C57BL/6 mice aged 8–12 weeks (n = 10) using sucrose density gradient centrifugation at 100,000 g, as described previously17. The extracted myelin mass was weighed by using a balance (Sartorius, Germany) with an accuracy of 0.001 g and diluted to a concentration of 100 mg/mL with sterile PBS. 1 μL Dil (Cell membrane red fluorescent staining kit) (Beyotime, China) were added to the 200 μL myelin stock solution and incubated at 37 °C for 1 h to obtain 200 μL fluorescent Dil-myelin with a concentration of 100 mg/mL.
Preparation of macrophage and myelin conditioned medium
RAW 264.7 cells (Purchased from the Cell Bank of the Chinese Academy of Sciences) were cultured in a six-well plate at a density of 2 × 105 cells/mL with normal DMEM containing 10% FBS for 24 h. Then, the cell culture medium was replaced with serum-free DMEM for another 24 h, and the supernatant was collected as the RAW macrophage conditioned medium after centrifugation at 2000 rpm for 15 min. Myelin conditioned medium was obtained by diluting the myelin stock solution (100 mg/mL) with DMEM to a concentration of 0.5 mg/mL. The medium was preserved at 4 °C and protected from light for a maximum period of 1 week.
Cell proliferation experiments
Cells were seeded in a 96-well plate at a density of 2000 cells/well. The hBcl2, cb, NC and BMSCs groups were established, with 4 parallel wells set for each group at each time point. After 1, 2, 3, 4, 5, and 6 days of cell culture, 100 μL fresh DMEM was replaced, and 2 μL of Cell Counting Kit-8 (CCK8) (Biosharp, China) solution was added to each well. The plate was then incubated at 37 °C for 4 h. At least three independent replicates were performed for the above experiment. The absorbance at 450 nm was measured using a microplate reader (Molecular Devices, USA).
Serum deprivation experiment
Cells were seeded in a 96-well plate at a density of 4000 cells/well. The hBcl2, NC, cb, and BMSCs groups were established, with 4 parallel wells set for each group at each time point. After culturing the cells with DMEM containing 10% FBS for 1 day, the DMEM medium without FBS was replaced to continue culturing the cells in each group. After 1, 2, 3, 4, 5, 6, 7 and 8 days of cell culture, CCK8 analysis was performed as mentioned before. At least three independent replicates were performed for the above experiment.
Western blot analysis
Western blot was performed to confirm the transfection efficiency of lentiviral vectors. Protein samples (20 µg) isolated from each cell group were loaded and separated via 10% sodium SDS-polyacrylamide gel electrophoresis, followed by transfer to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk in Tris-buffered saline with 0.5% Tween-20 (TBST) at room temperature for 30 min. After washing the membranes with TBST, they were incubated overnight at 4 °C with mouse anti-β-Tubulin (1:10,000, T0023, Affinity, USA) and mouse anti-hBcl2 antibody (1:100, NBP2-15200, Novus, USA). Then, the membranes were incubated with goat anti-mouse secondary antibody (1:10,000, A4416, Sigma, USA) conjugated with horseradish peroxidase at room temperature for 1 h. Labeled proteins were visualized on the membranes using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific, USA). Finally, Image J (NIH, USA) was used for quantitative analysis. The intensity of the β-Tubulin bands was used for normalization.
Immunocytochemistry
The cells were fixed in 4% paraformaldehyde for 5 min and blocked with 5% donkey serum albumin at room temperature for 30 min. They were then incubated with the primary antibodies at 4 °C overnight. The primary antibodies used for staining were as follows: mouse anti-hBcl2 (1:100, NBP2-15200, Novus, USA), rabbit anti-cleaved caspase3 (1:100, ab32042, Abcam, USA), and mouse anti-Lamp1 (1:100, sc20011, Santa Cruz, USA). The samples were incubated with secondary antibodies at room temperature for 1 h. The secondary antibodies used were as follows: donkey anti-mouse Alexa Fluor 488, donkey anti-mouse Alexa Fluor 647, donkey anti-mouse Alexa Fluor 594, and donkey anti-rabbit Alexa Fluor 594 (1:500, Termo Fisher Scientific, USA). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, C1005, Beyotime Biotechnology, China) was used for nuclear staining.
Establishment of the SCI animal model and BMSCs transplantation
Female SD rats were used in the animal experiments. The rats were randomly divided into three groups: (1) NC group (SCI + negative control BMSCs transplantation; n = 3); (2) hBcl2 group (SCI + hBcl2 overexpressing BMSCs transplantation; n = 3); (3) cb group (SCI + BMSCs overexpressing hBcl2-cb transplantation; n = 3). After anesthesia with 2% pentobarbital sodium, rats were subjected to laminectomy at the T9-10 vertebral level. Dumont forceps with a 0.4-mm-wide tip (11223-20, Fine Science Tools, Germany) were used to completely compress the spinal cord from both sides for 10 s, resulting in moderate compression injury18. Bladders were emptied manually twice daily, and the rats were allowed free feeding. Three days after SCI, the rats were anesthetized again, and their spinal cords were re-exposed. The cell suspension concentration were adjusted to 2 × 104/μL using a cell counter, and the cell suspension was slowly injected on both sides of the lesion epicenter using a siliconized Hamilton syringe (7634-01 and 7803-05, Hamilton, Switzerland)19,20. A volume of 5 μL of cell suspension was injected on each side at a rate of 0.5 μL/min, and the needle was held in place for 5 min before being slowly removed. The muscles and skin were sutured sequentially, and the rats were transferred to the animal house for culture after iodine sterilization.
Tissue preparation and immunofluorescence staining
Rats were anesthetized with 2% pentobarbital sodium, and the blood was removed by transcardial perfusion with PBS. A 5-mm segment of spinal cord tissue surrounding the injury site was harvested for histological analysis after perfusion with 4% paraformaldehyde. The spinal cord segment was then embedded in OCT embedding agent, and 16 μm sagittal sections were obtained using a microtome (RM2235, Leica, Germany). The sections were blocked in 10% donkey serum containing 0.3% Triton X-100 (SL050 and T8200, Solarbio, China) at room temperature for 1 h, followed by overnight incubation with primary antibodies at 4 °C. The primary antibodies included mouse anti-NF200 (1:500, ab213128, Abcam, USA), rabbit anti-GFAP (1:200, 16825-1-AP, Proteintech, China), mouse anti-hBcl2 (1:100, NBP2-15200, Novus, USA), and rabbit anti-cleaved caspase3 (1:100, ab32042, Abcam, USA). Then, the sections were incubated with secondary antibodies at room-temperature for 1 h. The secondary antibodies used were donkey anti-mouse Alexa Fluor 555, donkey anti-rabbit Alexa Fluor 488, and donkey anti-mouse Alexa Fluor 488, donkey anti- rabbit Alexa Fluor 555 (1:500, Termo Fisher Scientific, USA). Finally, the sections were stained with DAPI to label the nuclei. The images of the sections were acquired using a Zeiss LSM 900 confocal microscope system and a Zeiss Axio Scope A1 fluorescence microscope.
Statistical analysis
Data were presented as mean ± SD. All experiments were carried out independently at least 3 times. Quantification was conducted using a blind method. Statistical analysis was performed using Graph Pad Prism 5.0 (La Jolla, USA). The unpaired t test was used to analyze the data between two groups. Multiple comparisons were analyzed by one-way or two-way analysis of variance (ANOVA), followed by Tukey’s post hoc test. Values of P < 0.05 were considered to have significant differences.
Ethical approval
All animal experiments were carried out in accordance with institutional animal care protocols and were authorized by Anhui Medical University Ethical Committee (No. LLSC20160052). Adult female Sprague-Dawley (SD) rats were supplied from Experimental Animal Center of Anhui Medical University (No. SCXK (Wan) 2017-001).
Statement
The study is reported in accordance with ARRIVE guidelines.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41598-024-52167-4