All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise specified.
Culture of human mesenchymal stem cells (hMSCs)
HMSCs were isolated from bone marrow obtained as discarded tissue during routine surgery from four adult donors at the Lariboisiere Hospital, Paris, France. The tissues of interest were collected with the patient’s informed consent and agreed with Lariboisiere Hospital regulations. All ethical regulations relevant to human research participants were followed. HMSCs were isolated from each donor’s bone marrow using a procedure adapted from literature reports31,32, characterized by CD marker expression and differentiation potential, and cultured in Alpha Minimum Essential Medium (α-MEM; PAN Biotech GmbH, Aidenbach, Germany) supplemented with 10% Fetal Bovine Serum (FBS; PAA Laboratories GmbH, Les Mureaux, France) and 1% antibiotic/antimycotic (atb/atm; v/v, PAA Laboratories GmbH, Cörbe, Germany) under standard cell culture condition, that is, a humidified, 37 °C, 5% CO2, and 95% air environment. When 80–90% confluence was reached, the hMSCs were trypsinized using trypsin-EDTA (Sigma-Aldrich, St Quentin Fallavier, France) and replated at a density of 10×103 cells/cm². HMSCs from the four donors were pooled at an equal ratio at passage 1 and further expanded to passage 4 before use.
Transduction of hMSCs
The pooled hMSCs (at passage 2) were transduced with a lentiviral vector encoding the firefly luciferase and the ZSGreen proteins (pRRLsin-MND-Luc-IRES2-ZS Green-WPRE; TRANSBIOMED, Bordeaux, France) as previously described7 and further expanded. Flow cytometry analysis of ZSGreen-positive cells showed that 88% of the hMSCs were transduced. These cells will be referred to as “Luc-ZS Green-hMSCs” thereafter.
Culture of Human Umbilical Vein Endothelial Cells (HUVECs)
HUVECs (Lonza, Walkersville, USA) were cultured in Endothelial Cell Growth Medium (EBM-2; Lonza, Levallois-Perret, France) supplemented with 5% FBS and 1% ATB/ATM, under standard cell culture conditions. When 80–90% confluence was reached, the HUVECs were trypsinized and replated at a density of 15 × 103 cells/cm². Cells at passage 4 were used for the in vitro experiments.
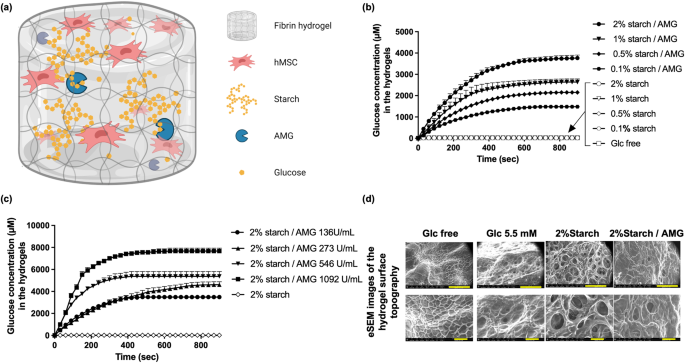
Preparation of wheat starch solutions
Wheat-derived starch solutions at either 0.2, 1, 2, or 4% (w/v) concentrations were prepared by stirring and heating starch powder in 10 mM HEPES buffer containing 0.3 M NaCl, 0.04 M CaCl2 (pH 7.4) at 92 °C for 2 h.
Preparation of hydrogels
Hydrogels were prepared by mixing two aqueous solutions (Mix 1 and Mix 2) detailed in Supplementary Table 1. Whenever relevant, hMSCs were added in the mix 1 before polymerisation. The Mix 1 was first deposited on a hydrophobic surface (specifically, a polytetrafluoroethylene (PTFE), disk diameter = 6 mm), and the Mix 2 was then added and homogenized within the Mix 1. These hydrogels were maintained at 37 °C in a humidified atmosphere for 1 h. After polymerization, each hydrogel was carefully removed from the PTFE surface, before use. Four types of cellularized hydrogels were prepared: (i) “Glc-free”: glucose-free hydrogel that represents a negative/empty control hydrogel; (ii) “Glc 5.5 mM”: glucose hydrogel loaded with 5.5 mM glucose that represents a standard control hydrogel; (iii) “Starch”: fibrin hydrogels loaded with starch to reach the final starch concentration of 0.1%, 0.5%, 1% or 2%; and (vi) “Starch/AMG”: fibrin hydrogels loaded with starch to reach the final starch concentration of 0.1%, 0.5%, 1% or 2% and amyloglucosidase (AMG). A concentration of 0.1% AMG was chosen because, of the AMG concentrations tested ranging from 136 U/ml to 1092 U/ml, a concentration of 136 U/ml AMG resulted in the slower degradation of starch in vitro. 100 µl hydrogels were loaded with 105 hMSCs for the in vitro experiments. 200 µl hydrogels were loaded with 2 × 105 Luc-ZS Green-hMSCs for the in vivo assessment of hMSC survival. 125 µl hydrogels were loaded with 1.25 × 105 hMSCs for the in vivo assessment of the proangiogenic potential of the hMSC secretome. For the in vitro culture experiments, hydrogels were placed in individual wells of 12-wells tissue-culture plates, and immersed in 2 mL serum- and glucose-free α-MEM culture medium and placed in a humidified incubator at 0.1% oxygen tension using a proOx-C-chamber system (C-Chamber, C-374, Biospherix, New York, NY).
Hydrogels surface topography
The surface topography of the hydrogels was assessed using environmental scanning electron microscopy (Philips eSEM FEG/XL-30). The specimens were treated with liquid nitrogen for 30 s just before scanning to make the external hydrogel surface visible and then photographed.
Hydrogel-contained glucose quantification without hMSCs
The amount of glucose inside the hydrogels was quantified using a glucose electrode biosensor (AniRA-Neurochem platform, University Lyon I, France) placed at the center of the samples33. The glucose microelectrode biosensor was composed of a 25 µm diameter platinum wire insulated in a pulled glass capillary with a 100 µm long sensing tip coated with electropolymerized poly-m-phenylenediamine and with glucose oxidase enzyme immobilized with poly(ethylene glycol) diglycidyl ether (PEGDE)33. Glucose oxidation into gluconolactone by the enzyme produced hydrogen peroxide (H2O2), which was reoxidized at the platinum electrode, creating an oxidation current directly proportional to glucose concentration (based on standard curve).
Near-anoxia culture conditions
The “ischemic” environment was simulated in vitro by culturing the hydrogel-contained hMSCs under both near-anoxia (0.1% O2) and nutrient (including both serum and glucose) deprivation17. Near-anoxia was achieved using a proOx-C-chamber system (C-Chamber, C-374, Biospherix, New-York, USA). The oxygen concentration in this chamber was maintained at 0.1%, with a residual gas mixture composed of 5% CO2 and balanced nitrogen at 37 °C for the experiment duration. Environmental nutrient deprivation was achieved using a glucose-free α-MEM (PAN Biotech GmbH).
Quantification of viable and apoptotic cells within hydrogels
The viability of hMSCs maintained inside the hydrogels under near anoxia was evaluated on day 1, 7, and 14 after trypsinization using Hoechst 33342 (HE) and propidium iodide (PI) staining and attune flow cytometer (Life Technologies, Saint Aubin, France)15. In details, at the prescribed times, the hydrogels were incubated with both 1 µg/mL nucleic acid stain Hoechst 33342 (HE; Sigma-Aldrich) and 1 µg/mL propidium iodide (PI; Sigma-Aldrich) at 37 °C for 20 min; the hydrogels were then observed and photographed using a Zeiss confocal microscope (LSM 800, Zeiss, Göttingen, Germany). Hydrogels were then digested, and hMSCs were detached from hydrogels using trypsin-EDTA for 20 min. Then PBS containing 2% bovine serum albumin (BSA, Sigma-Aldrich) was added to stop the chemical action of trypsin. After centrifugation (at 3,500xg for 5 min), the hMSCs were re-suspended in fresh PBS and analyzed using an Attune flow cytometer (Life Technologies, Saint Aubin, France). Cells staining both HE positive and PI negative were identified as “viable cells”, whereas those staining both HE positive and PI-positive were identified as “dead cells”. Cell viability was expressed as the number of viable cells at each point tested normalized with the respective viable cell number on day 1 of culture. For each hydrogel tested, cell viability on day 1 of culture was determined to be more than 90% of the 105 hMSCs seeded at day 0.
Proliferative potential of hydrogel-contained hMSC under near-anoxia
After 14 days under near-anoxia, hMSCs were trypsinized from the 2% starch/AMG hydrogels and transferred back into tissue culture-flask (103 cells/cm²) and standard cell culture conditions (i.e., 21% O2 and further cultured in α-MEM containing 5.5 mM glucose and 10% fetal bovine serum) for 10 days. The doubling time of these hydrogel-derived hMSCs was determined and compared to the one of naive hMSCs cultured under standard cell culture conditions for 14 days. The phenotype of these two hMSC population was also determined using flow cytometry as previously described17.
Preparation of hMSC conditioned media (CM)
The hydrogel-contained hMSCs were maintained under near anoxia with no cell culture medium change. On day 7 and 14, the supernatant cell culture media (further referred as conditioned media (CM)) were collected, centrifuged at 700 × g for 5 min, aliquoted, and kept at −80 °C until further use.
Assessment of the chemotactic effect of conditioned media
The chemo-attractive potential of CM from hydrogel-contained hMSCs maintained under near anoxia for either 7 or 14 consecutive days was determined using the Boyden chamber migration assay as described previously32. The hMSCs or HUVECs that had migrated through the porous membrane were photographed using a Keyence VHX-2000F microscope (Courbevoie, France) and counted using Image J free software (National Institute of Health, Bethesda, USA). In details, 600 µL of each respective conditioned media (CM) were added to the bottom well of the Boyden chamber, and 2 × 104 hMSCs or 5 × 104 HUVECs were seeded on the top of the 8-µm-pore-diameter porous membrane (disk diameter = 6.5 mm; Transwell®; VWR International, Fontenay-sous-Bois, France), previously coated with 0.5% gelatin (Sigma-Aldrich) and cultured in glucose- and serum-free media. Serum-free α-MEM and α-MEM supplemented with 10% FBS were added to the bottom wells as negative and positive controls. After 6 h of maintenance under standard cell culture conditions, the cells on the top of the porous membrane were scrapped to remove the hMSCs or HUVECs that had not migrated from the original seeding location. These porous membranes were then fixed using paraformaldehyde 11% (Sigma-Aldrich) at room temperature for 30 min and stained using an azure eosin methylene blue 0.4% solution (Giemsa; Sigma-Aldrich) for 3 min.
Assessment of released bioactive mediators
The presence of chemotactic and proangiogenic mediators in CM was quantified using Luminex technology (Millipore, Billerica, USA) following the manufacturer’s instructions32. The level of 16 mediators, specifically, IL-8, CCL2, MMP9, Chemerin, MIF, CXCL2, CXCL5, CXCL10 (known as chemotactic growth factors) and VEGF-A, VEGF-C, VEGF-D, ANGPT-1, ANGPT-2, ANG, FGF-BASIC, Endostatin (known as angiogenic modulators) was evaluated using the MasterPlex QT 1.0 system (MiraiBio, Alameda, USA) and analyzed using Luminex-100 software version 1.7 (Luminex, Austin, USA).
Animals
Ten-week-old female nude immunodeficient mice (NMRI-nu (nu/nu) were obtained from Janvier Labs (Le Genest-Saint-Isle, France). Animal experiments were conducted in accordance with the European Directive 2010/63/EU regarding the protection of animals used for scientific purposes and were approved by the Ethics Committee on Animal Research (APAFIS #14805-2018041119309138 v3).
Ectopic implantation
Before each surgical procedure, a dose of buprenorphine (Buprecare®; 0.1 mg/kg animal weight; Axience, Pautin, France) was administrated subcutaneously in each mouse, and the skin was prepared for surgery using an application of povidone-iodine (Betadine®, Vetoquinol, Paris, France). Anesthesia was induced by intraperitoneal administration of ketamine (Ketamine1000®; 100 mg/kg animal weight; Virbac, France) and xylazine (Rompun® 2%; 8 mg/kg animal weight; Bayer HealthCare, Berlin, Germany). Flowing oxygen was delivered using a specific mask for each animal throughout the surgical procedure. Either two (for the assessment of cell proangiogenic potential) or four (for the evaluation of cell viability) symmetrical incisions (each 7.5 mm in length) on both sides of the spine were made on the back of each mouse, and subcutaneous pouches were created. Hydrogels were then carefully and randomly inserted into each pouch. Skin closure was accomplished with an interrupted suture pattern using 4.0 polyglactin 910 sutures (Ethicon, Issy-les-Moulineaux, France). The mice were monitored daily by trained animal-care personnel throughout the postoperative period. Food and water were available ad libitum to the animals.
Assessment of AMG leakage from hydrogels
A fluorescently labelled AMG was prepared by coupling AMG with Dylight 800 N-hydroxysuccinimide-ester (Thermofischer Scientific) (AMGDL800). Briefly, a Recombinant Aspergilus Niger AMG powder (Megazyme) was resuspended in water and the buffer was exchanged against 0.05 M sodium borate pH 8.5 using a SpinOUT GT600 column (G-biosciences). 4.5 mg AMG in borate buffer was mixed with 250 µg of Dylight 800 NHS incubated at room temperature for 1 h and then dialyzed overnight at 4 °C against 0.1 M sodium citrate buffer pH 5.5. AMGDL800 protein concentration and degree of labelling was determined using a Spectramax ABS+ microplate reader and a spectradrop micro-volume microplate (Molecular Devices). Calculations were done according to the Dylight 800 manufacturer instructions, giving a 0.89 mol dye /mol protein. Hydrogels containing 60 µg of a mix of AMG and AMGDL800 in a ratio of 90/10 were then prepared as aforementioned. Before implantation, the area corresponding to each hydrogel was measured and used to delineate a region of interest beyond which AMG fluorescence was considered to have leaked from the hydrogel. Hydrogels were then implanted ectopically in mice. On days 0, 1,2,3,4,7,10, and 14, animals were anesthetized with isofluorane, and fluorescence efficiency in the region of interest was measured non-invasively using a fluorescence imaging system (Ivis, Lumina II, Caliper Life Sciences, Villebon-sur-Yvette, France) according to34; These data were normalized to those obtained on day 0 after surgery.
Assessment of cell viability
The viability of the Luc-ZS Green-hMSCs contained within the implanted hydrogels was monitored non-invasively using a bioluminescence imaging system on days 1, 7, and 14 (Ivis, Lumina II, Caliper Life Sciences, Villebon-sur-Yvette, France), as previously described35. For this purpose, the mice were anesthetized by delivery of 3% isoflurane (Iso-Vet®; Piramal HealthCare, Northumberland, UK) in oxygen, and 100 mg/kg D-luciferin was injected intramuscularly in the area of the hydrogel locations. The mice were then placed in a ventral position inside the detection chamber of the bioluminescence system and maintained under anesthesia. Images were taken every 5 min for 1 h. A region of interest surrounding each hydrogel was manually defined on each image, and the photon flux emitted by each hydrogel was quantified using the Living Image® 3.2 software (Caliper Life Sciences). The highest BioLuminescence Intensity (BLI) signal was retained for each mouse and time point tested. The percentage of viable cells post-implantation was determined as the photon fluxes measured at each time point tested normalized with the respective BLI signal acquired the day after surgery (i.e., day-1 of data acquisition). On days 7 and 14, the mice were sacrificed (using an overdose of intracardiac pentobarbital delivery (Dolethal®; Vetoquinol, Paris, France), and the hydrogels were explanted, fixed in 4% paraformaldehyde, and embedded in paraffin. All prepared paraffin sections were processed for human beta-2-microglobulin immunodetected (hβ2-MG, a membrane protein that stains human cells) using the Envision+ kit (Dako, Les Ulis, France) and a polyclonal rabbit anti-hβ2-MG (1/1000, Novocastra, Nanterre, France) as the primary antibody as previously described36.
Assessment of the proangiogenic potential of hMSCs-containing-hydrogels
Each 125 µl hydrogel (loaded with 1.25 × 106 hMSC) was first placed in the center of a silicone tube (Silicone DIA; 4 mm inner diameter, 6 mm outer diameter, 16 mm height; Weber Métaux, France) and then sandwiched between two layers (3 mm height) of fibrin gel added at each tube end. The latter gel, composed of 1 mg/mL fibrin without aprotinin, was completely degraded 24 h after implantation. Blood vessels were visualized by injecting the mice with a radio-opaque polymer compound (Microfil®, Flowtech, Carver, MA, USA) at 14 or 21 days post-implantation. For this purpose, each animal was deeply anesthetized, and the skin from the thorax and the rib cage was incised to access the heart. The left ventricle was catheterized using a 20 G cannula (BD Venflon, Beckson Dickinson Infusion, Sweden), and the right atrium was cut for blood removal. Each animal was first perfused with isotonic saline (50 mL) containing heparin (100 UI/mL) using a pump (at a 6 mL/min flow rate) for 6 min to rinse the blood from the vasculature. 14 mL of Microfil® (prepared according to the manufacturer’s instructions using 6.3 mL of Microfil®, 7 mL of the specific diluent, and 0.7 mL of the specific curing agent) were then manually perfused at approximately 2 mL/min to force the Microfil® into the capillary networks without extravasation into the surrounding tissue. The perfused euthanized animals were stored at 4 °C overnight to allow polymerization of the Microfil®; the silicone tubes containing the hydrogels were explanted and fixed in 4% paraformaldehyde overnight. The specimens were then imaged using a Skyscan1172 micro-CT-scanner (Bruker, Evere, Belgium) with voltage 80 kV, current 100 mA, exposure for 85 ms, and 0.3-degree rotation step settings without any filter. The images obtained had 10 μm pixel size. The scanned images were then reconstructed as a stack of slices of each sample using Nrecon software, 16 bits (Bruker, Evere, Belgium). The volumes of interest (VOI; 2 per sample) were set as cylinders overlapping the internal diameter of the silicone tube (4 mm), between the 1st and the 3rd mm height from the top and the bottom of the silicone tube edges. Indeed, as some hMSC-containing hydrogels tested partly resorbed after implantation, the aforementioned method allowed to define a similar volume of interest for each sample rigorously. New blood vessel volume was reported as the amount of binarized object volume measured within the designated volume of interest within the threshold gray values 120–255 on CTan software (Bruker). Values regarding new blood vessel thickness and numbers were calculated using the abovementioned thresholds. The new blood vessel length was calculated from the formula V = h*pi*r2 on the assumption that the new blood vessel diameter was homogeneous.
Statistics and reproducibility
Numerical data were expressed as means + standard of the mean (SEM). Statistical analyses were performed using commercially available software (GraphPad v9.3.1 Software, California Corporation, USA). In vitro experiments were conducted with at least 3 biological replicates per group whereas in vivo experiments were conducted using at least 5 biological replicates for each group of hydrogels tested. The effects of hydrogel group, time of culture/implantation, and their interactions were examined using two-way ANOVAs. For each significant effect, a Tuckey’s post-hoc test was conducted. When only two groups of hydrogels were compared at a single time point of analysis (i.e., Supplementary Fig. 1b), a t-test with a Welsh’s correction was conducted. For all analyses, a p-value < 0,05 was considered significant. For a given group, the significance of the difference between experimental times of culture/implantation are indicated by the α symbol; the significance between groups at the same time of culture/implantation are indicated with p < 0.05. *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s42003-023-05643-y