AAV6 vector design, production and purification
AAV6 vector plasmids were cloned into the pAAV-MCS plasmid (Agilent Technologies) comprising inverted terminal repeats derived from AAV2. Gibson Assembly Master Mix (New England Biolabs) was used for the creation of all DNA repair vectors as per the manufacturer’s instructions. AAV6 vector was produced and purified with little variation from previously described processes41. 293T cells (Life Technologies) were seeded in five dishes (15 cm2) with 13 × 106–15 × 106 cells per plate at 24-h pretransfection. Each dish was then transfected with a standard polyethylenimine (PEI) transfection of 6 μg inverted-terminal-repeat-containing plasmid and 22 μg pDGM6 (gift from David Russell, University of Washington), which holds the AAV6 cap, AAV2 rep and Ad5 helper genes. After a 48–72-h incubation, cells were collected and vectors were purified using the AAVpro purification kit (catalogue number 6666; Takara Bio) per the manufacturer’s instructions and then stored at −80 °C until further use. AAV6 vectors were titred using ddPCR to measure the number of vector genomes as previously described42.
In vitro culture of CD34+ HSPCs
Human CD34+ HSPCs were cultured in conditions as previously described13,43,44,45,46. CD34+ HSPCs were isolated from cord blood (provided by Stanford Binns Program for Cord Blood Research) or sourced from plerixafor- and/or G-CSF-mobilized peripheral blood (AllCells and STEMCELL Technologies). Frozen plerixafor- and/or G-CSF-mobilized peripheral blood of patients with SCD were provided by Dr Vivien Sheehan at Emory University. CD34+ HSPCs were cultured at 1 × 105–5 × 105 cells ml−1 in StemSpan Serum-Free Expansion Medium II (STEMCELL Technologies) or Good Manufacturing Practice Stem Cell Growth Medium (SCGM; CellGenix) supplemented with a human cytokine (PeproTech) cocktail: stem cell factor (100 ng ml−1), thrombopoietin (100 ng ml−1), Fms-like tyrosine kinase 3 ligand (100 ng ml−1), interleukin-6 (100 ng ml−1), streptomycin (20 mg ml−1), penicillin (20 U ml−1) and 35 nM of UM171 (catalogue number A89505; APExBIO). The cell incubator conditions were 37 °C, 5% CO2 and 5% O2.
Electroporation-aided transduction of cells
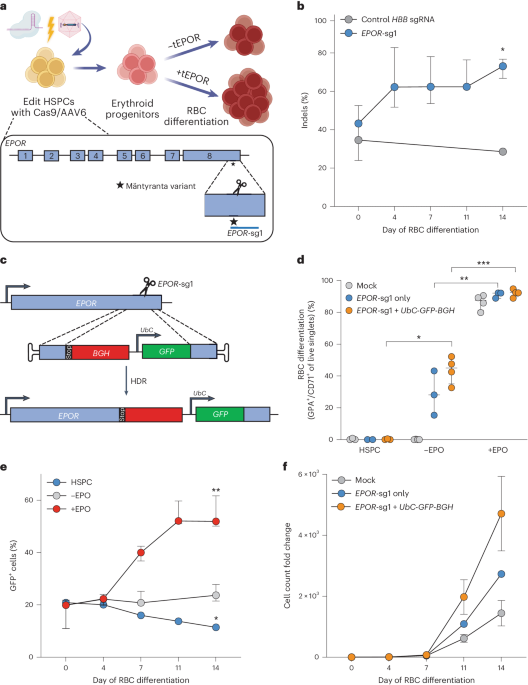
The synthetic chemically modified sgRNAs used to edit CD34+ HSPCs were purchased from Synthego or TriLink Biotechnologies and were purified by HPLC. These modifications comprise 2′-O-methyl-3′-phosphorothioate at the three terminal nucleotides of the 5′ and 3′ ends described previously17. The target sequences for the gRNAs were as follows.
EPOR gRNA (EPOR-sg1)
5′-AGCTCAGGGCACAGTGTCCA-3′
EPOR gRNA (EPOR-sg2)
5′-GCTCCCAGCTCTTGCGTCCA-3′
CCR5 gRNA (CCR5-sg3)
5′-GCAGCATAGTGAGCCCAGAA-3′
HBA1 gRNA (HBA1-sg4)
5′-GGCAAGAAGCATGGCCACCG-3′
The HiFi Cas9 protein was purchased from Integrated DNA Technologies (IDT) or Aldevron. Before electroporation, RNPs were complexed at a Cas9/sgRNA molar ratio of 1:2.5 at 25 °C for 10–20 min. Next, CD34+ cells were resuspended in P3 buffer (Lonza) with complexed RNPs and subsequently electroporated using the Lonza 4D-Nucleofector and 4D-Nucleofector X Unit (program DZ-100). Electroporated cells were then plated at 1 × 105–5 × 105 cells ml−1 in the previously described cytokine-supplemented media. Immediately after electroporation, AAV6 was dispensed onto cells at 2.5 × 103–5 × 103 vector genomes per cell based on titre determined by ddPCR. For multiplex editing experiments, in addition to the steps described above, cells were incubated with 0.5 μM of the DNA-PKcs inhibitor AZD7648 (catalogue number S8843; Selleck Chemicals) for 24 h, as previously described32,33.
Allelic modification analysis using ddPCR
Edited HSPCs were collected within 2–3 days postelectroporation and at each media change throughout erythrocyte differentiation and then analysed for modification frequencies of the alleles of interest. To quantify editing frequencies, we created custom ddPCR primers and probes to quantify HDR alleles (using in–out PCR and probe corresponding to the expected integration event) compared with an established genomic DNA reference (REF) at the CCRL2 locus14. QuickExtract DNA extraction solution (catalogue number QE09050; Biosearch Technologies) was used to collect genomic DNA input, which was then digested using BamHI-HF or HindIII-HF as per the manufacturer’s instructions (New England Biolabs). The percentage of targeted alleles within a cell population was measured with a Bio-Rad QX200 ddPCR machine and QuantaSoft software (v.1.7; Bio-Rad) using the following reaction mixture: 1–4 μl genomic DNA input, 10 μl of ddPCR Supermix for Probes (no dUTP; Bio-Rad), primer and probes (1:3.6 ratio; IDT), and volume up to 20 μl with H2O. ddPCR droplets were then generated following the manufacturer’s instructions (Bio-Rad): 20 μl of ddPCR reaction, 70 μl of droplet generation oil and 40 μl of droplet sample. Thermocycler (Bio-Rad) settings were as follows: 98 °C (10 min), 94 °C (30 s), 55.7–60 °C (30 s), 72 °C (2 min), return to step 2 for 40–50 cycles and then 98 °C (10 min). Analysis of droplet samples was then performed using the QX200 Droplet Digital PCR System (Bio-Rad). We next divided the copies per microlitre for HDR (%): HDR/REF. The following primers and probes were used in the ddPCR reaction.
CCR5 (for tEPOR–YFP construct)
Forward primer (FP): 5′-GGGAGGATTGGGAAGACA-3′
Reverse primer (RP): 5′-AGGTGTTCAGGAGAAGGACA-3′
Probe: 5′-6-FAM/AGCAGGCATGCTGGGGATGCGGTGG/3IABkFQ-3′
HBA1 (for tEPOR–YFP construct)
FP: 5′-AGTCCAAGCTGAGCAAAGA-3′
RP: 5′-ATCACAAACGCAGGCAGAG-3′
Probe: 5′-6-FAM/CGAGAAGCGCGATCACATGGTCCTGC/3IABkFQ-3′
HBA1 (for HBB construct and tEPOR-HBB constructs)
FP: 5′-GTGGCTGGTGTGGCTAATG-3′
RP: 5′-CAGAAAGCCAGCCAGTTCTT-3′
Probe: 5′-6-FAM/CCTGGCCCACAAGTATCACT/3IABkFQ-3′
HBA1 (for HBB-tEPOR constructs)
FP: 5′-TCTGCTGCCAGCTTTGAGTA-3′
RP: 5′-GCTGGAGTGGGACTTCTCTG-3′
Probe: 5′-6-FAM/ACTATCCTGGACCCCAGCTC/3IABkFQ-3′
CCRL2 (reference)
FP: 5′-GCTGTATGAATCCAGGTCC-3′
RP: 5′-CCTCCTGGCTGAGAAAAAG-3′
Probe: 5′-HEX/TGTTTCCTC/ZEN/CAGGATAAGGCAGCTGT/3IABkFQ-3′
Indel analysis using TIDE software
Within 2–4 days postelectroporation, HSPCs were collected with QuickExtract DNA extraction solution (catalogue number QE09050; Biosearch Technologies) to collect genomic DNA. The following primer sequences were used to amplify the respective cut sites at the EPOR locus:
FP: 5′-CAGCTGTGGCTGTACCAGAA-3′
RP: 5′-CAGCCTGGTGTCCTAAGAGC-3′
Sanger sequencing of the respective samples was then used as input for indel frequency analysis relative to a mock, unedited sample using TIDE as previously described25.
In vitro differentiation of CD34+ HSPCs into erythrocytes
Following editing, HSPCs derived from healthy individuals or patients with SCD were cultured for 2–3 days as described above. Subsequently, a 14-day in vitro differentiation was performed in supplemented SFEMII medium as previously described24,47. SFEMII base medium was supplemented with 100 U ml−1 penicillin–streptomycin, 10 ng ml−1 SCF (PeproTech), 1 ng ml−1 IL-3 (PeproTech), 3 U ml−1 EPO (eBiosciences), 200 μg ml−1 transferrin (Sigma-Aldrich), 3% human serum (heat-inactivated; Sigma-Aldrich or Thermo Fisher Scientific), 2% human plasma (isolated from umbilical cord blood provided by the Stanford Binns Cord Blood Program), 10 μg ml−1 insulin (Sigma-Aldrich) and 3 U ml−1 heparin (Sigma-Aldrich). Cells were cultured in the first phase of medium for 7 days at 1 × 105 cells ml−1. In the second phase of medium, days 7–10, cells were maintained at 1 × 105 cells ml−1 and IL-3 was removed from the culture. In the third phase of medium, days 11–14, cells were cultured at 1 × 106 cells ml−1, with a transferrin increase to 1 mg ml−1.
Immunophenotyping of differentiated erythrocytes
Differentiated erythrocytes were analysed by flow cytometry on day 14 for erythrocyte lineage-specific markers using a FACS Aria II (BD Biosciences). Edited and unedited cells were analysed using the following antibodies: hCD45-V450 (HI30; BD Biosciences), CD34-APC (561; BioLegend), CD71-PE-Cy7 (OKT9; Affymetrix) and CD235a-PE (GPA) (GA-R2; BD Biosciences). In addition to cell-specific markers, cells were also stained with Ghost Dye Red 780 (Tonbo Biosciences) to measure viability.
Haemoglobin tetramer analysis
Frozen pellets of approximately 1 × 106 in vitro-differentiated erythrocytes were thawed and lysed in 30 µl of RIPA buffer with 1× Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) for 5 min on ice. The mixture was vigorously vortexed and cell debris was removed by centrifugation at 13,000 r.p.m. for 10 min at 4 °C. HPLC analysis of haemoglobins in their native form was performed on a cation-exchange PolyCAT A column (35 mm2 × 4.6 mm2, 3 µm, 1,500 Å; PolyLC) using a Perkin-Elmer Flexar HPLC system at room temperature and detection at 415 nm. Mobile phase A consisted of 20 mM Bis-Tris and 2 mM KCN at pH 6.94, adjusted with HCl. Mobile phase B consisted of 20 mM Bis-Tris, 2 mM KCN and 200 mM NaCl at pH 6.55. Haemolysate was diluted in buffer A before injection of 20 µl onto the column with 8% buffer B and eluted at a flow rate of 2 ml min−1 with a gradient made to 40% B in 6 min, increased to 100% B in 1.5 min, returned to 8% B in 1 min and equilibrated for 3.5 min. Quantification of the area under the curve of the peaks was performed with TotalChrom software (Perkin-Elmer) and raw values were exported to GraphPad Prism 9 for plotting and further analysis.
mRNA analysis
After differentiation of HSPCs into erythrocytes, cells were collected and RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). Subsequently, cDNA was made from approximately 100 ng of RNA using the iScript Reverse Transcription Supermix for quantitative PCR with reverse transcription (Bio-Rad). Expression levels of the β-globin transgene and α-globin mRNA were quantified with a Bio-Rad QX200 ddPCR machine and QuantaSoft software (v.1.7; Bio-Rad) using the following primers and 6-FAM/ZEN/IBFQ-labelled hydrolysis probes, purchased as custom-designed PrimeTime qPCR Assays from IDT.
HBB and HBB-tEPOR into HBA1
FP: 5′-GGTCCCCACAGACTCAGAGA-3′
RP: 5′-CAGCATCAGGAGTGGACAGA-3′
Probe: 5′-6-FAM/AACCCACCATGGTGCATCTG/3IABkFQ-3′
To normalize for RNA input, levels of the RBC-specific reference gene GPA were determined in each sample using the following primers and HEX/ZEN/IBFQ-labelled hydrolysis probes, purchased as custom-designed PrimeTime qPCR Assays from IDT.
GPA (reference)
FP: 5′-ATATGCAGCCACTCCTAGAGCTC-3′
RP: 5′-CTGGTTCAGAGAAATGATGGGCA-3′
Probe: 5′-HEX/AGGAAACCGGAGAAAGGGTA/3IABkFQ-3′
ddPCR reactions were created using the respective primers and probes and droplets were generated as described above. Thermocycler (Bio-Rad) settings were as follows: 98 °C (10 min), 94 °C (30 s), 54 °C (30 s), 72 °C (30 s), return to step 2 for 50 cycles and then 98 °C (10 min). Analysis of droplet samples was done using the QX200 Droplet Digital PCR System (Bio-Rad). To determine relative expression levels, the numbers of HBB transgene copies per millilitre were divided by the numbers of GPA copies ml−1.
Methylcellulose CFU assay
At 2–3 days postelectroporation, HSPCs were plated in SmartDish 6 well plates (catalogue number 27370; STEMCELL Technologies) containing MethoCult H4434 Classic or MethoCult H4434 Classic without EPO (catalogue numbers 04444 and 04544; STEMCELL Technologies). After 14 days, the wells were imaged using the STEMvision Hematopoietic Colony Counter (STEMCELL Technologies). Colonies were counted and scored to determine the number of CFU-GEMM (colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte), CFU-GM (colony-forming unit-granulocyte, macrophage), BFU-E (burst-forming unit-erythroid) and CFU-E (colony-forming unit-erythroid) colonies.
Quantification of editing efficiency at evaluated off-target sites
Potential sgRNA off-target sites were predicted using the CRISPR Off-target Sites with Mismatches, Insertions and Deletions (COSMID) online tool48. Sites were ranked according to score and duplicate predictions at the same location were removed. All sites with a score ≤5.5 were included in the analysis and the 5 sites in exonic or untranslated regions were further analysed. PCR amplification of these sites was performed using genomic DNA from mock-edited and RNP-edited cells. The following primers were used with Illumina adaptors (FP adaptor, 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′; RP adaptor, 5′-GACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′).
EPOR-OT1
FP: 5′-GAGCGGGCTACAGAGCTAGA-3′
RP: 5′-TGGCAGAAAGTAAGGGGATG-3′
EPOR-OT2
FP: 5′-ACTTGTGGAGCCACAGTTTG-3′
RP: 5′-AATGCCCTTGAGATGAATGC-3′
EPOR-OT3
FP: 5′-TCACACACCCGTAGCCATAA-3′
RP: 5′-AGAATGCTCTTTGCAGTAGCC-3′
EPOR-OT4
FP: 5′-CTCAAAACTTCACCCAGGCT-3′
RP: 5′-GGTCTGTCATTGAATGCCTT-3′
EPOR-OT5
FP: 5′-CAACCCTGATGGGTCTGC-3′
RP: 5′-CCACAGCTGGCTGACCTT-3′
Following amplification, PCR products were purified by gel electrophoresis and subsequent extraction using the GeneJet Gel Extraction Kit (catalogue number FERK0692; Thermo Fisher Scientific). Purified samples were submitted for library preparation and sequencing by Amplicon-EZ next-generation sequencing (Azenta Life Sciences), ensuring a yield of over 100,000 reads per sample. Amplicons, flanked by Illumina partial adaptor sequences, which encompassed the programmed DSBs for CRISPR–Cas9, underwent sequencing using Illumina chemistry. FastQC (v.0.11.8, default parameters; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to assess the quality of raw reads. Subsequently, paired-end reads were aligned to the specified off-target regions using CRISPResso2 (v.2.2.14; fastq.gz files were used as input)49.
Statistical analysis
GraphPad Prism 9 software was used for all statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41551-024-01222-6