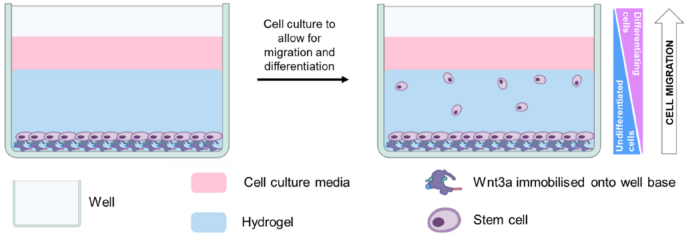
Wnt3a immobilization
Recombinant mouse Wnt3a protein was purchased from R&D Systems and stock solutions, prepared by reconstituting the protein to 10 µg/ml in a solution of 1% CHAPS buffer (Merck), were stored in aliquots at − 80 °C for up to 3 months. Wnt3a working solutions were prepared by diluting the stock solution to 600 ng/ml in Dulbecco’s Phosphate Buffered Solution (DPBS) (Gibco). Wnt3a was immobilized on the surface of 96-well plate (Corning) by adding 40 µl of protein working solution in previously modified wells. Well plate modification was performed by adding a 2% (3-Aminopropyl)triethoxysilane (APTES) (Merck) solution in 90% ethanol (Merck) and incubating for 30 min at room temperature (RT), followed by two washed with 100% ethanol. After 1 h of incubation at RT, Wnt3a solution was removed, followed by a wash with DPBS. For control wells, Wnt3a protein was inactivated after immobilization with a freshly made 20 mM Dithiothreitol (DTT) (Merck) solution in DPBS, which was incubated at 37 °C for 30 min and washed with DPBS. In order to block exposed silane groups, prior to cell culture all wells were covered for 1 h in basic medium, consisting of high glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco), 1% L-Glutamine solution (Merck) and 1% penicillin/streptomycin solution (PS) (Merck).
Y201 cell culture and characterization
Cell culture
Y201 bone-marrow stromal cells (BMSCs), kindly donated by Prof. Paul Genever from the University of York, were stably transfected with 7xTCF-GFP/SV40-mCherry as previously described.26,29 Cells were routinely passaged every 2–3 days in basic medium when they reached ~ 80% confluency. For all experiments cells were used up to passage 20. For chondrogenic differentiation (also referred to as ‘TGF-β3’ condition), medium consisted of high glucose DMEM supplemented with 1% FBS, 1% L-glutamine, 1% PS, 10% sodium pyruvate solution, 40 μg/ml L-proline, 50 μg/ml L-ascorbic acid-2-phosphate, 10% insulin-transferrin-selenium solution (ITS), 100 nM dexamethasone (all from Merck) and 10 ng/ml recombinant human TGF-β3 (Peprotech). Kartogenin-supplemented medium was prepared using high glucose DMEM supplemented with 5% FBS and 100 nM kartogenin (Selleck Chemicals).
Cells were pelleted and differentiated into chondrocytes using a high-throughput v-bottomed 96-well plate culture system, following previously established protocols30. Each well of an autoclave-sterilized v-bottomed 96-well polypropylene microplate (Greiner bio-one) received 200,000 cells at passage 3, which were subsequently centrifuged at 500 × g for 5 min. The resulting cell pellets were cultured in 250 µl of chondrogenic differentiation medium, composed of high glucose DMEM supplemented with 2 mM L-glutamine, 100 U/mL penicillin-0.1 mg/ml streptomycin, 100 μg/mL sodium pyruvate, 40 μg/mL L-proline, 50 μg/mL L-ascorbic acid-2-phosphate, 4.7 μg/mL linoleic acid, 1.5 mg/mL bovine serum albumin (BSA), 1 × insulin-transferrin-selenium, 100 nM dexamethasone (all obtained from Merck), and 10 ng/ml recombinant human TGF-β3 (Peprotech). Throughout the experiment, the culture medium was refreshed three times weekly, and the cell pellets were maintained for a total duration of three weeks.
In order to investigate Y201 cell response to Wnt3a, cells were detached, counted and seeded on 96-well plates at a density of at approximately 5 × 104 cells/cm2. Cell were cultured for 4 days in basic medium with/without 50 ng/ml Wnt3a.
Histology
Samples of Y201 cell pellets in basic and chondrogenic conditions (n = 2 per group) were fixed in 10% neutral buffered formalin for 2 h and embedded in paraffin. Samples were cut as 5 µm-thick slices and deposited on SuperFrost™ glass slides (ThermoScientific). Sections were de-waxed in xylene for 5 min, followed by re-hydration in descending grades of ethanol to water. Histological slides were stained with Toluidine blue (Merck) for production of proteoglycans and GAGs, and counterstained with Gill’s number 2 Hematoxylin (Merck) according to manufacturer’s instructions. Sections were then washed in absolute ethanol thrice and cleared in xylene before mounting with DPX mountant (Merck). Sections were imaged using an EVOS XL Core microscope.
Assessment of gene expression
Y201 cell pellets in basic and chondrogenic conditions (n = 3 per group) were snap frozen after 26 days of culture and homogenized using disposable pellet pestles (Merck). RNA was then extracted using TRI Reagent (Merck) and converted into cDNA using high-capacity cDNA Reverse Transcription Kits (Applied Biosystems) (both as per the manufacturer’s instructions). Gene expression analysis was performed for ACAN and COL2A1 using SYBR Green-based quantitative real-time polymerase chain reaction (qRT-PCR) with pre-optimized QuantiTect primer assays (Qiagen) and an AriaMx Real-Time PCR System (Agilent Technologies). qRT-PCR data were analyzed using the ΔΔCt method as described previously31, with gene expression normalized to the reference gene GAPDH.
Wnt3a/Gel platform preparation
Preparation of hydrogel precursor solutions
For gellan gum hydrogels, a 1% (w/v) solution was prepared by dissolving Phytagel™ (Merck) in dH2O. The solution was heated in a microwave for 5 s, followed by manual swirling for 2–3 s and this process was repeated until the powder was completely dissolved (~ 3 min). The precursor solution was UV-sterilized for 90 s and then incubated at 37 °C for at least 30 min to allow for cooling without setting and maintained at 37 °C until needed.
For collagen hydrogels, stock rats tail collagen type I solution (Corning) was diluted to 1 mg/ml using basic medium supplemented with 20% HEPES buffer. The hydrogel solution was maintained in ice to avoid setting of the gel until needed.
For Poly(ethylene glycol) (PEG) hydrogels, a 5% (w/v) solution was prepared by dissolving Poly(ethylene glycol) diacrylate pellets (ThermoFisher) in DPBS. Then, Irgacure® 1173 (BASF) photoinitiator was added (1 µl in 1 ml of PEG solution) and the precursor solution was left in the dark until needed.
For GelMA hydrogels, polymer synthesis was performed as previously described.32 Briefly, a 10% (w/v) solution was prepared by dissolving gelatine type A from porcine skin (300 bloom strength, Merck) in DPBS at 60 °C for 30 min. Methacrylic anhydride (MA) (Merck) was added to the gelatine solution to a final concentration of 6% (v/v) and left to react for 2 h at 50 °C under vigorous stirring. After the reaction period, the solution was transferred to 50 mL tubes and the unreacted MA was partially removed by centrifugation at 3500 rpm for 5 min at RT. The solution was then dialyzed against distilled water for 7 days at 37 °C using 12–14 kDa cut-off dialysis tubes (Thermo Scientific). After dialysis, the GelMA solution was diluted to 2% (w/v) and the pH adjusted to 7.4 using 1 mM sodium hydroxide solution (Merck). Lastly, GelMA solutions were lyophilized for 2 days to generate a white porous foam, which was stored at − 80 °C until further use. After GelMa synthesis, a 4% (w/v) hydrogel precursor solution was prepared by dissolving the synthesized polymer in DPBS at 60 °C for 30 min and UV-sterilized for 5 min. Then, 40 mM phosphated riboflavin (Merck) and 546 mM sodium persulphate (Merck) solution were prepared in dH2O and filter-sterilized. The two photoinitiators were added into the GelMA solution at a final concentration of 2 mM and 10 mM for riboflavin and sodium persulfate, respectively, and maintained in the dark until needed.
Set-up of the 3D model
Y201 cells were detached, resuspended in serum-free medium and seeded onto Wnt3a-immobilized 96-well plates at a density of 3 × 104 cells per well. Cells were left to adhere for at least 1 h before the medium was removed and the cells were covered with 50 µl per well of the appropriate hydrogel solution. To allow for gelation, gellan gum and collagen precursor solutions were incubated for 10 min at 37 °C, while PEG samples were irradiated with broad spectrum UV light for 5 min at a distance of 1.7 cm. GelMA precursor solution was set using a 100 mW/cm2 Knightsbridge FLF Floodlight visible light lamp (RS Components) for 5 min at a distance of 5.5 cm. After the hydrogels were set, 200 µl of appropriate medium was added on top of the gels and samples were incubated at 37 °C and 5% CO2 for 1 h, before the medium was replaced. Basic, chondrogenic or kartogenin-supplemented medium (described above in Section “Cell culture“) was used for samples in basic, ‘TGF-β3’ and ‘kartogenin’ conditions, respectively. After 72 h of culture, kartogenin-supplemented medium was replaced with high glucose DMEM supplemented with 5% FBS, 1% L-Glutamine, 1% PS and 1% ITS. For all conditions, during the culture period medium was replaced every other day.
Optical coherence tomography (OCT)
4% (w/v) GelMA hydrogels were prepared in 96-well plate as described in Section “Preparation of hydrogel precursor solutions“. Prepared hydrogels were covered with culture medium to simulate the experimental conditions and underwent OCT imaging on days 0, 3, and 7. A 20 µl medium was added while doing the imaging in order to prevent dehydration and reflection. Following the imaging procedure, 200 µl of the appropriate culture medium was added onto the hydrogels, and the samples were subsequently incubated at 37 °C. Mechanical property analysis was carried out utilizing debiased ambient vibration Optical Coherence Elastography (OCE). This technique, designed for mechanical contrast evaluation, was conducted using an OCT imaging system, with no supplementary hardware modifications. This approach allows for the non-contact, sterile assessment of mechanical properties, relying on using ambient vibrations. The OCT system employed in this study featured a lateral scanning distance of approximately 3.7 mm. Mechanical property assessment encompasses the generation of mechanical contrast maps and quantitative analysis through the calculation of Young’s modulus (E). E is derived using a calibration curve established through nanoindentation and OCT imaging. The correlation between E and the estimated wavelength (λ) follows the equation E ∝ λ2, which justifies the utilization of the mean wavelength squared, λ2, in E calculation33.
Biochemical assays
Y201 cells cultured in monolayer (n = 4 per group) were washed with DPBS, detached and centrifuged to form a pellet, while the 3D cell constructs (n = 4 per group) were only washed with DPBS. 250 µL of Proteinase K (Invitrogen) (1 mg/mL dissolved in 100 mM ammonium acetate, pH 7.0) was added per 100 mg wet weight of sample. Samples were vortexed, then incubated at 60 °C with an additional vortex every 30 min until complete dissociation had occurred (~ 2 h). Proteinase K was inactivated by heating the samples to 100 °C for 5 min in a heat block. DNA content was quantified using the PicoGreen dye assay (Biosciences) following the kit instructions with a calf thymus DNA standard. Sulfated glycosaminoglycan (sGAG) content within the sample was quantified using the dimethylmethylene blue (DMMB) dye-binding assay with a chondroitin sulfate standard. The sGAG content secreted into media was analyzed using Glycosaminoglycan Assay Blyscan kit (Biocolor) following the kit instruction, also with a chondroitin sulfate standard. Cell proliferation in monolayers was quantified using the EdU (Abcam) and Ki67 (Abcam) assays following the kit instructions.
Immunofluorescence staining
Y201 cells were either cultured in monolayers at a seeding density of approximately 5 × 104 cells/cm2 or 5 × 104 cells/ml in a 96-well plate for 7 days or in 3D hydrogels for 7–14 days, as described above. Samples were fixed in 10% neutral buffered formalin for 15 min (cell monolayers) or 2 h (cells in hydrogels) and simultaneously permeabilized and blocked with 2% bovine serum albumin (BSA)/0.1% Triton-X in PBS for 2 h. Cells were stained for STRO-1 (Abcam), SOX9 (Abcam), NCAM (R&D Systems) and COL2A1 (Merck). All primary antibodies were diluted in 1% BSA/0.1% Triton-X at a working concentration of 10 µg/ml and incubated overnight at 4 °C. After washing with PBS, cells were labelled with donkey anti-rabbit AlexaFluor488 (ThermoFisher), donkey anti-goat AlexaFluor568 (ThermoFisher) and/or donkey anti-mouse AlexaFluor 647 (ThermoFisher) diluted at a working concentration of 2 µg/ml in 1% BSA/0.1% Triton-X and incubated for 2 h at RT. Cells were washed with PBS thrice, counterstained with DAPI (Merck) for 15 min and stored in PBS. Fluorescence microscopy was performed on an EVOS M5000 microscope for cell monolayers, while a confocal Olympus IX81 microscopewas used to image 3D hydrogels. Based on the number of migrated cells, the migration results were categorized. If there were < 10 cells, this was categorized as poor migration as that was a relatively low proportion of the total in the bottom. If more than 10 cells were observed, this was categorized as average migration, while migration that exceeded 400 µm in the 500 µm analysis was considered as good migration.
Statistical analysis
Statistical analysis was performed using GraphPadPrism software package (Dotmatics). Unpaired t-test was used to compare between two groups. Statistical significance was considered if p < 0.05. Data are presented as mean ± standard deviation (SD).
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41598-024-65970-w