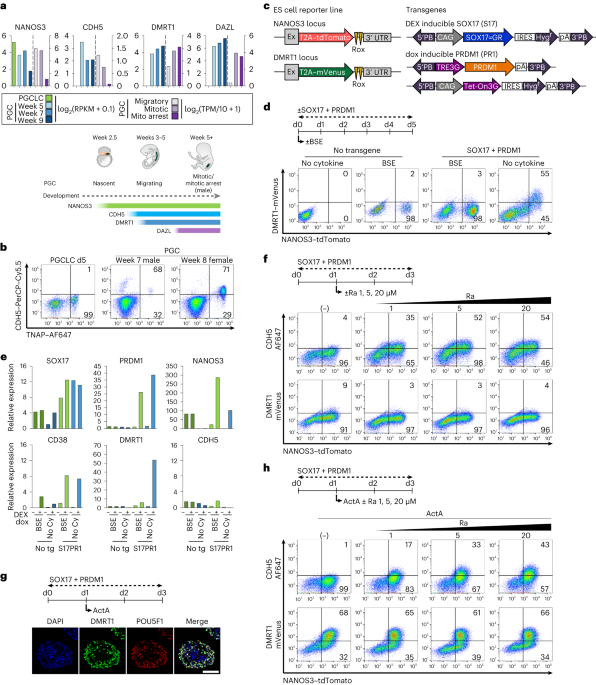
Ethics statement
Human embryonic tissues were used with permission from the National Health Service Research Ethical Committee, UK (Research Ethics Committee number 96/085). Patients (who had already decided to undergo the termination of pregnancy operation) fully and freely consented to donate the foetal tissues for medical and academic research. We received genital ridges and dissected to isolate gonads from mesonephric tissues. The gonadal tissues were dissociated into single-cell suspension with Collagenase IV (2.6 mg ml−1) (Sigma, C5138) and DNase I (10 U ml−1) in Dulbecco’s modified Eagle medium (DMEM)–F/12 (Gibco). Cells were resuspended in fluorescence-activated cell sorting (FACS) medium (phosphate-buffered saline (PBS) with 3% foetal calf serum) with 5 μl of Alexa Fluor 488 anti-alkaline phosphatase (BD Pharmingen, 561495) and 5 μl of PerCP–Cy5.5 anti-CDH5 (BD Pharmingen, 561566) antibodies for flow cytometry. Medical or surgical termination of pregnancy was carried out at Addenbrooke’s Hospital, Cambridge, UK. This study did not involve the use of human gametes, pre-implantation embryos or experimental models mimicking early human development. Where applicable, our study is compliant with the International Society for Stem Cell Research guidelines. All samples were handled and stored according to the Human Tissue Act regulations. The Gurdon Institute safety committee carried out appropriate scrutiny, including risk assessments.
Collection of PGCs from human embryos
Crown–rump length and anatomical features, including limb and digit development, were used to determine the developmental stage of human embryos with reference to Carnegie staging. The sex of embryos was determined by sex determination PCR as previously described90. Genital ridges were dissected and separated from surrounding mesonephric tissues and dissociated into single-cell suspension with Collagenase IV (2.6 mg ml−1) (Sigma, C5138) and DNase I (10 U ml−1) in DMEM–F/12 (Gibco) at 37 °C for 15–30 min. Cells were resuspended in FACS medium (PBS with 3% foetal calf serum) with 5 μl of Alexa Fluor 488 anti-alkaline phosphatase (BD Pharmingen, 561495) and 5 μl of PerCP–Cy5.5 anti-CDH5 (BD Pharmingen, 561566) antibodies for 20 min at room temperature. Flow cytometry was performed with BD LSRFortessa Cell Analyzer (BD Biosciences), and dot plots were generated by FlowJo software.
Cell culture
Approval for the use of all ES cell lines used in this study was granted by the MRC Steering Committee for the UK Stem Cell Bank and for the Use of Stem Cell Lines. Male ES cell line, WIS2 (46XY), was kindly provided by Weizmann Institute of Science, Israel91. Female ES cell line, Shef-6 (46XX), was obtained from the UK Stem Cell Bank (UKSCB accession no. R-05-031). 4i ES cells were maintained on irradiated mouse embryonic fibroblasts (MEFs) (purchased from MTI-GlobalStem or prepared in house) in knockout DMEM (Thermo Fisher Scientific) supplemented with 20% knockout serum replacement, 0.1 mM non-essential amino acids, 0.1 mM 2-mercaptoethanol, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin, 2 mM l-glutamine, 20 ng ml−1 human LIF (Stem Cell Institute, University of Cambridge (SCI)), 8 ng ml−1 bFGF (SCI), 1 ng ml−1 TGFβ (Peprotech), 3 µM GSK3i (CHIR99021, Miltenyi Biotec), 1 µM ERKi (PD0325901, Miltenyi Biotec), 5 µM p38i (SB203580, TOCRIS Bioscience) and 5 µM JNKi (SP600125, TOCRIS Bioscience), as reported14. Cells were passaged every 2–4 days using TrypLE Express (Thermo Fisher Scientific). Before seeding 4i ES cells on MEFs, 10 µM of ROCKi (Y-27632, TOCRIS Bioscience) was added into the medium. Conventional ES cells were maintained on vitronectin (Thermo Fisher Scientific)-coated plates in Essential 8 medium (Thermo Fisher Scientific) according to the manufacturer’s protocol. Cells were passaged every 3–5 days using 0.5 mM ethylenediaminetetraacetic acid (EDTA)/PBS.
To induce PGCLCs, 4i ES cells or preME (see below) cells were trypsinized into single cells and seeded into Corning Costar Ultra-Low attachment multiwell 96-well plates (Sigma) or AggreWell Microwell Plates (Stemcell Technologies) at 4,000–8,000 cells per well. PGCLC induction medium based on aRB medium contains 500 ng ml−1 BMP2 (SCI), 100 ng ml−1 SCF (Peprotech), 50 ng ml−1 EGF (R&D Systems) and 10 µM ROCKi. aRB medium is composed of Advanced RPMI 1640 Medium (Thermo Fisher Scientific) supplemented with 1% B27 supplement (Thermo Fisher Scientific), 0.1 mM non-essential amino acids, 100 U ml−1 penicillin–0.1 mg ml−1 streptomycin and 2 mM l-glutamine13. For DM+PGCLC induction, PGCLC induction medium was replaced with aRB medium containing 100 ng ml−1 ActA (SCI), 20 µM Ra (Sigma), 100 ng ml−1 SCF (Peprotech) and 50 ng ml−1 EGF (R&D Systems) as indicated. For preME induction, trypsinized ES cells cultured in E8 were seeded on vitronectin-coated dish at 200,000 cells per well in 12-well plates in preME induction medium that is composed of aRB medium supplemented with 100 ng ml−1 ActA (SCI), 3 µM GSK3i and 10 µM ROCKi. For induction of exogenous transgenes, 100 µM DEX (Sigma) and/or 1 µg ml−1 dox (Sigma) was added.
Vector construction and transfection
For construction of reporter knock-in targeting vector, 5′ and 3′ arms amplified from human genomic DNA and combined with tdTomato or mVenus and Rox–PGK–PuroΔtk–Rox were cloned into modified NANOS3–tdTomato targeting vector containing MC1-promoter-driven diphtheria toxin A using in-fusion HD cloning kit (Takara Bio)13. Guide RNAs targeting around the stop codon sequence of DMRT1 or DAZL genes (Supplementary Table 1) were cloned into pX330 (Addgene). For construction of dox-inducible system, DMRT1 and BCL2L1 were cloned into PiggyBAC pCMV–Tet3G vector used previously13. All fragments were amplified by PCR using PrimeSTAR MAX, PrimeSTAR GXL DNA polymerase (Takara Bio) or Q5 High-Fidelity DNA Polymerase (NEB) according to the manufacturer’s protocol.
Plasmid transfection for gene targeting or transgene introduction was carried out with electroporation or lipofection as described before13,14. In brief, electroporation was carried out using Gene Pulser equipment (Bio-Rad) with 1–5 × 106 4i ES cells mixed with targeting vector and pX330 plasmid containing guide RNA. For lipofection, reverse transfection was carried out with 2 × 105 4i ES cells in 100–200 µl of Opti-MEM containing plasmid vectors and Lipofectamine 2000 or Lipofectamine Stem Transfection Reagent (Thermo Fisher) with 5 min incubation at room temperature. After electroporation or lipofection, ES cells were seeded onto 4 drug resistant (DR4) MEFs (GlobalStem or SCI) and 48 h later, 0.5 µg ml−1 puromycin (Sigma) or 25 µg ml−1 hygromycin B (Thermo Fisher Scientific) was added to the culture medium for selection. Drug-resistant ES cell colonies were picked up and genotyped for correct targeting by PCR using primers in Supplementary Table 1. The targeted clones were expanded and then used for excision of Rox-flanked PGK–PuroΔtk by transient transfection of pCAG–Dre–IH. After selection with 25 µg ml−1 hygromycin B and subsequently with 0.2 µM fialuridine, colonies were picked up and assessed for excision by PCR using primers in Supplementary Table 1 (Extended Data Fig. 1c).
qPCR
Total RNA was extracted using PicoPure RNA Isolation Kit (Thermo Fisher) and cDNA was synthesized using QuantiTect Reverse Transcription Kit (QIAGEN) according to manufacturer’s protocols. RT–qPCR was performed using QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher). Primer sequences are listed in Supplementary Table 3. Values shown were normalized to housekeeping genes and relative changes to control sample values.
Genomic DNA was extracted using Quick-DNA Microprep Plus Kit (Zymo). Primer sequences for genomic DNA quantification are listed in Supplementary Table 4. Values shown were normalized to human genomic locus for TPOX and normalized to wild-type sample copy numbers.
Immunofluorescence and image analysis
Aggregates were fixed in 4% paraformaldehyde for 1–2 h at 4 °C and embedded in OCT compound (VWR) for frozen sections. Sections were incubated with primary antibodies for 1–2 h at room temperature or overnight at 4 °C and with fluorescent-conjugated secondary antibodies (dilution 1:500) for 1 h at room temperature. Primary antibodies are listed in Supplementary Table 5 (anti-DMRT1, rabbit, monoclonal, Abcam, cat. no. ab166893, dilution 1:500; anti-POU5F1, mouse, monoclonal, BD Biosciences, cat. no. 611203, dilution 1:500; anti-DAZL, rabbit, polyclonal, Abcam, cat. no. ab34139, dilution 1:200; anti-5mC, rabbit, monoclonal, Cell Signaling Technology, cat. no. 28692, dilution 1:200; anti-5mC, mouse, monoclonal, Abcam, cat. no. ab10805, dilution 1:150; anti-5hmC, rabbit, polyclonal, active motif, cat. no. 39769, dilution 1:500; anti-DNMT3B, sheep, polyclonal, R&D Systems, cat. no. AF7646, dilution 1:200; anti-TFAP2C, rabbit, polyclonal, Santa Cruz Biotechnology, cat. no. sc-8977, dilution 1:200; anti-SOX9, goat, polyclonal, R&D Systems, cat. no. AF3075-SP, dilution 1:200; anti-tdTomato, goat, polyclonal, SICGEN, cat. no. AB8181, dilution 1:100; anti-DDX4, rabbit, monoclonal, Abcam, cat. no. 235442, dilution 1:200; anti-mitochondria, mouse, monoclonal, Abcam, cat. no. ab92824, dilution 1:800; anti-SOX17, goat, polyclonal, R&D Systems, cat. no. AF1924, dilution 1:100; APC conjugated SUSD2, mouse, monoclonal, BioLegend, cat. no. 327408, dilution 1:100; anti-TFCP2L1, goat, polyclonal, R&D Systems, cat. no. AF5726, dilution 1:100). After antibody treatment, sections were stained with 4′,6-diamidino-2-phenylindole (Sigma) and imaged using Leica SP8 inverted laser scanning confocal microscope by white laser. HC PL APO CS2 63× 1.4 numerical aperture oil immersion objective was used. Image analyses were performed using a custom script92 for Fiji93, which segments nuclei in 4′,6-diamidino-2-phenylindole channel with difference of Gaussian threshold using Otsu’s method94 and measures intensity in channels for 5mC, 5hmC.
Flow cytometry analysis
Aggregates were trypsinized with trypsin/EDTA (0.25%, Thermo Fisher) at 37 °C for 5–15 min and single-cell suspension was incubated with Alexa Fluor 488 or 647 conjugated anti-alkaline phosphatase (TNAP) antibody (BD Bioscience, 5 µl per sample), PerCP–Cy5.5-conjugated anti-CDH5 antibody (BioLegend, 5 µl per sample) and/or Alexa Fluor 647 conjugated anti-CD38 antibody (BioLegend, 5 µl per sample) and analysed using BD LSRFortessa Cell Analyzer (BD Bioscience). Flow cytometry data were analysed using FlowJo software.
Luciferase assay
For vector construction of luciferase assay, three genomic regions with DMRT1 binding peaks containing DMRT1 motif (hg38; peak 1: chr3:16,608,590–16,608,949, DMRT1 motif: aaaactatgttact; peak 2: chr3:16,602,880–16,603,116, DMRT1 motif: aatacatagtagta; peak 3: chr3:16,594,400–16,597,625 DMRT1 motif: ttgatacaatgttt) in day 4 DZ+PGCLCs at DAZL locus were amplified from human genomic DNA. These sequences were cloned into a piggyBAC-based luciferase (Luc+) reporter plasmid containing a hygromycin-resistant gene driven by a PGK promoter using in-fusion HD cloning kit. DMRT1 motif is scanned using HOMER scanMotifGenomeWide.pl function. The sequences without DMRT1 motif were amplified from the original plasmid with each peak’s sequences using the primers listed in Supplementary Table 2. ALR/alpha consensus sequences (aattctcagtaacttccttgtgttgtgtgtattcaactcacagagttgaacgatcctttacacagagcagacttgaaacactctttttgtggaatttgcaagtggagatttcagccgctttgaggtcaatggtagaataggaaatatcttcctatagaaactagacagaat, DMRT1 motif sequence: ttgaaacactctttt) were downloaded from Repbase. The synthesized ALR oligos from Merck were cloned into a piggyBAC-based luciferase (Luc+) reporter plasmid containing a hygromycin-resistant gene driven by a PGK promoter using in-fusion HD cloning kit.
HEK 293 cells (ATCC CRL-1573) were transfected using Lipofectamine 2000 Transfection Reagent (Thermo Fisher) with a piggyBAC plasmid containing a constitutively expressed green fluorescent protein (GFP) cassette and a neomycin-resistant cassette, a piggyBAC plasmid containing a dox-inducible DMRT1 transgene and a puromycin-resistant cassette, and a plasmid encoding a piggyBAC transposase. Following 4 days of ±dox treatment, cells were measured for GFP with Hidex Sense (HIDEX) and subjected to luciferase activity assay using the Dual-Glo Luciferase Assay System (Promega). Normalized luciferase activities were obtained by dividing firefly luciferase activity by GFP signal counts.
Western blot
Nuclear proteins were extracted using EpiQuik Nuclear Extraction Kit II (EPIGENTEK) and were separated on a Novex 4–20% Tris-Glycine Mini Gel (Thermo Fisher) using XCell SureLock Mini-Cell Electrophoresis System (Thermo Fisher) and transferred to Hybond P 0.45 µm polyvinylidene fluoride membrane (GE Healthcare). After blocking in 5% skimmed milk, the membrane was incubated with primary antibodies (anti-SOX17, rabbit, monoclonal, Cell Signaling Technology, cat. no. 81778, dilution 1:1,000; anti-PRDM1, rabbit, monoclonal, Cell Signaling Technology, cat. no. 9115, dilution 1:500; anti-DMRT1, rabbit, monoclonal, Abcam, cat. no. ab126741, dilution 1:1,000; anti-LaminB1, rabbit, polyclonal, Abcam, cat. no. ab16048, dilution 1:1,000; Supplementary Table 6). The antibody binding was detected by horseradish-peroxidase-conjugated anti-rabbit IgG (Dako; dilution 1:2,000 in 0.01% TBST) in conjunction with the Western Detection System (GE Healthcare).
Preparation of scRNA-seq libraries
Reporter or cell surface marker-positive cells were sorted by BD FACSAria III Cell Sorter and loaded according to the manufacturer’s protocol for the Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 (Dual Index) (10x Genomics) to attain between 2,000 and 6,000 cells per reaction. Library preparation was carried out according to the manufacturer’s protocol. Libraries were sequenced, aiming at a minimum coverage of 40,000 raw reads per cell, on the Novaseq 6000 systems using the sequencing format: read 1, 28 cycles; i7 index, 10 cycles; i5 index, 10 cycles; read 2, 90 cycles.
Preparation of bulk RNA-seq libraries
RNA-seq library was generated with total RNA (300 ng) using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760, NEB) with NEBNext rRNA Depletion Kit v2 (NEB) according to manufacturer’s protocol. Library was quantified using NEBNext Library Quant Kit Quick Protocol (E7630, NEB). Libraries were sequenced for 150 cycles in paired-end mode on the NovaSeq platform.
C&R
C&R for DMRT1 and normal rabbit IgG was performed as described43,44,45. Briefly, 50,000 purified DZ+PGCLCs were washed and bound to activated 10 μl Concanavalin A-coated magnetic beads. The beads were then incubated with wash buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM spermidine and protease inhibitor) containing 0.1% digitonin and 1 μg of DMRT1 antibody (ab126741, Abcam) or normal rabbit IgG (#2729, Cell signaling) for 2 h at 4 °C on a rotator. After two washes in digitonin–wash buffer, beads were resuspended in Protein A/G-MNase fusion protein at 70 ng ml−1 in digitonin–wash buffer and incubated for 1 h at 4 °C on a rotator. After two washes in digitonin–wash buffer (the beads with replicate 3 of day 4 DZ+PGCLC and day 8 DZ+PGCLC were washed with low-salt rinse buffer (20 mM HEPES, pH 7.5, 0.5 mM spermidine and 0.1% digitonin) once additionally), beads were resuspended in ice-cold calcium incubation buffer (3.5 mM HEPES pH 7.5, 10 mM CaCl2 and 0.1% digitonin). After 15 min, 2× stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM egtazic acid, 0.1% digitonin, RNase A 100 μl ml−1 and glycogen 50 μg ml−1) was added. Beads were incubated at 37 °C for 30 min, the liquid was removed to a fresh tube and DNA was extracted with phenol–chloroform extraction.
DNA library preparation and sequencing
Sequencing libraries were prepared with the NEBNext Ultra II DNA Library Prep Kit (NEB, E7645S) for Illumina according to the manufacturer’s protocol but without size selection and PCR enrichment of adaptor-ligated DNA. PCR enrichment of adaptor-ligated DNA was performed with KAPA HiFi Real-Time PCR Library Amplification Kit (Roche, KK2702) following the manufacturer’s recommendations. The number of PCR cycles using the KAPA polymerase was 7–10. SPRIselect beads (Beckman Coulter, B23317) were used for clean-up PCR product and size selection. Libraries were sequenced for 150 cycles in paired-end mode on the NovaSeq platform.
TAPS with βGT blocking and chemical-assisted pyridine borane sequencing plus
TAPS with βGT blocking (TAPSβ) and chemical-assisted pyridine borane sequencing plus (CAPS+) were performed according to previous publications47,48. Briefly, DNA was spiked with spike-in control DNA and sonicated to 300–500 bp, before ligation with NEBNext Adaptor for Illumina using KAPA HyperPrep Kit according to the manufacturer’s protocol. The uracil in the loop of NEBNext Adaptor was removed by USER Enzyme (New England Biolabs). A total of 100 ng ligated DNA was used for both TAPSβ and CAPS+. For TAPSβ, the ligated library was subjected to βGT (Thermo Fisher) blocking, two rounds of mTet1 oxidation, and borane reduction. For CAPS+, the ligated library was subjected to chemical oxidation and borane reduction. Converted DNA from TAPSβ and CAPS+ was amplified with NEBNext Multiplex Oligos for Illumina and KAPA HiFi HotStart Uracil+ ReadyMix PCR Kit for four cycles according to the manufacturer’s protocol. The PCR product was purified with Ampure XP beads. Libraries were sequenced for 150 cycles in paired-end mode on the NovaSeq 6000 platform.
Data processing for scRNA-seq
The reads were demultiplexed and aligned to the 10x Genomics’ GRCh38-2020-A reference genomes using the Cell Ranger Software (v.7.0.0, 10x Genomics) with default parameters. The summary statistics from Cell Ranger is provided in Supplementary Table 7.
We employed Scrublet to identify and distinguish single cells from cell doublets in each individual library. As described in ref. 95, we used a two-step diffusion doublet identification followed by Bonferroni–false discovery rate (FDR) correction and a significance threshold of 0.01. We used Scanpy v.1.8.0 (ref. 96) to analyse the filtered count matrices that were generated by Cell Ranger, following their recommended standard practices. Specifically, we excluded genes that were expressed by fewer than three cells and excluded cells that expressed fewer than 3,000 genes or had more than 10% mitochondrial content. We then normalized the raw counts by library size and log-transformed them. Next, we identified the highly variable genes, which we used for principal components analysis (PCA). We corrected for the library effect using Harmony97 on the PCA space (default parameters except theta = 1). Finally, we used the Harmony-corrected PCA space to identify the k (k = 15) nearest neighbours, perform Leiden clustering and visualize the results using UMAP. Leiden clusters with overall high doublet score or low counts number were flagged and discarded in further analysis. We used Seurat’s v.4.0.5 FindAllMarkers() function to identify up- and downregulated genes in each library with |log2fold change (FC)| >1 (ref. 98). To determine the cell cycle phase (that is, G1, S or G2/M) of each cell, we combined the expression of G2/M and S phase markers and used the method implemented in Scanpy’s score_genes_cell_cycle function to classify the cells99. We then compared the in vitro cell states identified in our study with the in vivo cell states reported in the Smart-seq2 dataset of gonadal cells from Li et al.24 (GSE86146). To do this, we downloaded the normalized transcripts per million (TPM) matrix from Li et al.24 and annotated their cells using the ‘FullAnnot’ field. We only considered the male foetal germ cell clusters. We used the tool scmap100 to project the Li et al.24 annotations onto our dataset and visualized the results of the projections using a dot plot.
Data processing for bulk RNA-seq
Trim Galore101 was used to remove the low-quality reads and adaptor sequences. Trimmed sequence files were mapped to human reference genome (GENCODE, GRCh38.p13) and counts on genes were generated using STAR102 with parameters –outFilterMultimapNmax 1 –outFilterMatchNmin 35. Normalized counts (normalize the total number of mapped reads per experiment to 1 × 108) on repeat elements were generated with the analyzeRepeats.pl of the HOMER103 package. Differential gene (or repeat element) expression analysis was performed with the glm method of the edgeR104 package for protein-coding genes. DEGs or repeat elements were identified with fold changes greater than 2 and FDR smaller than 0.05. Reads per kilobase of transcript per million mapped reads (RPKM) values of genes were calculated using Cufflinks105.
Secondary data analyses were performed using Microsoft Excel and R software version 4.0.5 with the packages ggplot2. GSEA106 was performed using the GSEA software by the Broad Institute. GO analysis was performed on the basis of GO Biological Process (http://geneontology.org). Marker protein-coding genes, 142 for migratory, 288 for mitotic and 937 for mitotic arrest male PGCs, were used on the basis of published markers identified from single cell RNA-seq data24,28, ‘PGC genes’ were identified on the basis of shared enriched DEGs (logFC >1, FDR <0.05) between week 7 and week 9 male PGCs against week 7 gonadal somatic cells or conventional ES cells6.
Data processing for C&R
To trim the short fragments that are frequently encountered in C&R experiments we used leeHom package program107 with —ancientdna option. The trimmed reads were aligned to the human reference genome (GENCODE, GRCh38.p13) using Bowtie2 2.2.6 (ref. 108) with options –very-sensitive –no-mixed –no-discordant -q –phred33 -I 10 -X 700. For MACS2 peak calling, parameters used were macs2 (ref. 109) callpeak –keep-dup all and the peaks with −log10(q value) >10 for day 4 DZ+PGCLC and the peaks with −log10(q value) >8 with IgG as control for day 8 DZ+PGCLC were selected. A total of 11,920 (day 4) and 7,818 (day 8) peaks that are in common between the replicates were used for further analysis. Peaks were annotated to their nearest genes or overlapping repeat elements using Homer annotatePeaks.pl function. To analyse the enriched TF motifs over peaks or repeat elements, HOMER findMotifsGenome.pl function was used.
Data processing for TAPSβ and CAPS+ methylome
The reads were demultiplexed using i7 sequences. The total sequencing reads number and conversion rate are provided in Supplementary Table 8. Trim Galore was used to remove the low-quality reads, and Samtools rmdup function was used to remove PCR duplicates. Trimmed reads were mapped to human reference genome (GENCODE, GRCh38.p13), and modified bases were called by asTair110. The methylation rate (%) for each CpG was calculated as the ratio between T and (C + T). Average CpG methylation levels of annotated genomic regions were calculated using UCSC bigWigAverageOverBed considering only information from CpGs with >5× coverage. To identify DMRs, we used DMRfinder111 with the default setting except –meanDiff_cutoff (5mC, 0.2; 5hmC, 0.05) and –pctMinCtrl 0 –pctMinExp 0 as sets of CpGs with a t-statistic greater than the critical value for α = 0.05 and with a gap <300 bases.
Statistics and reproducibility
For RNA-seq, C&R and 5hmC/5mC methylome data, two independent biological replicates (except for day 4 DZ+PGCLC C&R with three independent biological replicates) were included according to the guidelines of the Encode Consortium101. No statistical method was used to pre-determine sample size in other experiments. Low-quality replicates of libraries were excluded from the analysis, as determined by percentage of reads in peaks, number of peaks and genome browser visualization. As all results involved equipment-based quantitative measure and no subjective rating of data was involved, blinding and randomization are not relevant. All the data met the assumptions of the statistical tests used, including whether normality and equal variances were formally tested. All the data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Automotive / EVs, Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- ChartPrime. Elevate your Trading Game with ChartPrime. Access Here.
- BlockOffsets. Modernizing Environmental Offset Ownership. Access Here.
- Source: https://www.nature.com/articles/s41556-023-01224-7