This research was performed in accordance with all relevant ethical regulations; Mice were treated in accordance with a protocol approved by the Rockefeller University Institutional Animal Care and Use Committee.
Plasmids and lentivirus production
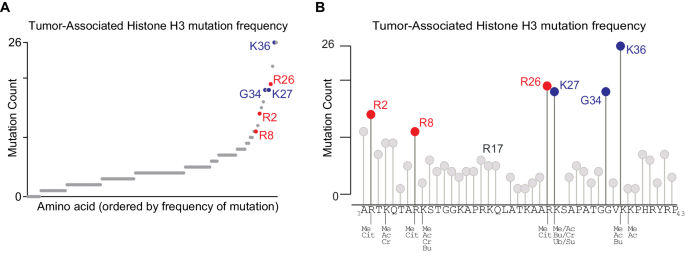
The human H3C2 cDNA sequence was cloned into the pCDH-EF1-MCS-puro lentiviral vector with C-terminal HA- and FLAG-epitope tags13. This construct was then used as a substrate for PCR-based site-directed mutagenesis using the Q5 site-directed mutagenesis kit (New England Biolabs) following the manufacturer’s protocol to generate histone point mutants H3.1 R2C, R2A, R2H, R8C, R8A, R8H, R17C, R17A, R17H, R26C, R26A, R26A, R26H. The presence of the correct mutations was confirmed by Sanger sequencing. To generate lentiviral particles, HEK293T cells were transfected with the pCDH-EF1-MCS-puro lentiviral vector harboring the H3.1 insert of interest along with helper plasmids (psPAX2 and pVSVG). The virus containing supernatant was collected in standard culture media containing 1% BSA on day two and filtered.
Cell lines, cell culture, and generation of stable cell lines
HEK293T and C3H10T1/2 (Clone 8, CCL-26, ATCC) cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) with 10% fetal bovine serum (CellGro) and 1% penicillin/streptomycin (Gibco). V6.5 mouse ES cells (C57BL/6 × 129S4/SvJae F1)43 (mESCs) were grown on gelatin-coated tissue cultures dishes in Knockout DMEM (Gibco) with 15% ES qualified FBS (Sigma), 0.072% beta-mercaptoethanol, 2mM L-glutamine (Gibco), MEM non-essential amino acids (from 100X stock; Gibco), and LIF (GeminiBio). Cell lines were tested for mycoplasma contamination. To generate cell lines expressing transgenic H3.1, cells were transduced with lentivirus with 5 µg/mL polybrene (Millipore). After 48 h, transduced cells were grown under selection with puromycin (2 µg/mL).
Arginine methyltransferase inhibitor treatment
Cells were seeded at 1 × 106 cells per 15 cm dish and allowed to recover overnight. The following day, the cells were treated with 300 nM MS023 (Selleck, S81112), 300 nM GSK591 (Selleck, S8111), 300 nM MS023 and 300 nM GSK591, or vehicle (DMSO). The final DMSO concentration was 0.5% for all conditions. After 2 days of treatment, the cells were collected, washed in PBS, and flash frozen for further analysis.
Immunoblotting
Protein samples were separated by SDS-PAGE and transferred to a PVDF membrane (or to nitrocellulose for blots of whole cell extracts with anti-methylarginine or beta-actin antibodies and and for anti-H3K4me3 from histone extracts), which was subsequently blocked with 5% milk solution in tris-buffered saline with 0.1% Tween-20 (TBST). Membranes were then probed with primary antibodies in 1% milk (or 5% BSA for anti-methylarginine and beta-actin blots, and for H3K27me3 blots from histone extract) in TBST overnight at 4 degrees Celsius, washed 3X with TBST and incubated with horseradish peroxidase-conjugated secondary antibody for detection using a chemiluminescent substrate (ECL, Pierce). Primary antibodies used were: anti-H3 (Abcam, ab1791; 1:3000-25000), anti-H3K4me3 (Active Motif, 39159; 1:1000), anti-H3K9me3 (Abcam, ab8898; 1:1000), anti-H3K27me3 (Cell Signaling, 9733; 1:1000-3500), anti-H3K27ac (Active Motif, 39133; 1:1000), anti-H3K27me1 (Active Motif, 61015; 1:1000), anti-H3K27me2 (Cell Signaling, 9728; 1:1000), anti-H3K4me1 (Abcam, ab8895; 1:1000), anti-H3K4me2 (abAbcam, 7766; 1:1000), anti-HA (Biolegend, 901503; 1:1000-5000), anti-SDMA (Cell Signaling, 13222; 1:1000), anti-ADMA (Cell Signaling, 13522; 1:1000), anti-MMA (Cell Signaling, 8015; 1:1000), and anti-beta-actin, (Cell Signaling, 4970; 1:1000). Secondary antibodies: anti-mouse IgG, HRP-linked (Cyvita, NA931; 1:5000), anti-rabbit IgG, HRP-linked (Dako, PO399; 1:5000), anti-rabbit IgG, HRP-linked (Cell Signaling, 7074; 1:2000-3000).
Histone acid extraction
Flash frozen pellets of 1-2 × 106 PBS-washed cells were thawed on ice and incubated on a rotator for 30 min at 4 °C in 1 mL of hypotonic lysis buffer (10 mM Tris-HCl, pH 8.0, 1 mM KCl, 1.5 mM MgCl2, 1 mM DTT, and 1x protease inhibitor cocktail (Roche)). Nuclei were pelted by centrifugation at 10,000 × g at 4 °C, resuspended in 0.4 N H2SO4 then incubated on a rotator at 4 °C for 30 min. The samples were then centrifuged at 16,000 × g at 4 °C for 10 min. The supernatant was then incubated with 132 µL trichloroacetic acid (100%), which was added in a dropwise fashion, for 30 min at 4 °C. The samples were then centrifuged at 16,000 × g at 4 °C for 10 min and the supernatant was discarded. The pellet was washed twice with 1 mL of ice-cold acetone, air-dried, and then resuspended in ddH20.
Nucleosome immunoprecipitation
Cell pellets of 3 × 107 HEK293T cells expressing transgenic epitope tagged histone H3.1 were collected and resuspended in 1 mL of LB1 (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton-X-100) with 1 × protease inhibitor cocktail (Roche). After a 10-min incubation on a rotator at 4 °C, tubes were spun at 1350 × g at 4 °C and the supernatant was discarded. The pellet was resuspended in 1 mL of LB2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) with 1 × protease inhibitors and incubated on a rotor at 4 °C for 10 min. After a 5-min spin at 1350 × g at 4 °C, the supernatant was discarded and the pellet was resuspended in 900 µL of LB3 (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-laurolysarcosine) with 1 × protease inhibitors and 100 µL of 10% Triton-X-100. In series, the sample was passed through 21 G, 23 G, and 27 G needles on ice to produce a homogenous mixture, which was then sonicated using a Covaris E220 ultrasonicator at a peak power of 220, duty factor of 5.0, and cycle/burst ratio of 200. After a 10-min spin at maximum speed in a 4 °C benchtop microcentrifuge, the supernatant was collected and transferred to tubes containing FLAG-conjugated magnetic beads (Pierce A36797) that had been equilibrated with PBS with 1 × protease inhibitor cocktail. Following an overnight incubation at 4 °C on a rotator, the supernatant was removed and the beads were washed three times with 900 µL LB3 with 1 × protease inhibitor cocktail and 100 µL of 10% Triton-X-100. Three additional washes with Buffer D (20 mM HEPES pH 7.9, 10% glycerol, 0.2 mM EDTA, 0.2% Triton-X-100 and 1 × protease inhibitor cocktail) with 100 mM NaCl and then three washes with PBS with 1 × protease inhibitor cocktail were performed prior to resuspending the beads in 100 µL of 100 mM glycine pH 2.0 and shaking at 1400 rpm at room temperature to elute the bound proteins (repeated for two elutions total). The eluate was then neutralized by vortexing with 15 µL of 1 M Tris-HCl pH 8.5.
Middle down histone sample preparation
Elutions from the FLAG- immunoprecipitation were dried down in a speed-vac, and then resuspended in 5 mM dithiothreitol (Thermo Fisher Scientific) in 50 mM ammonium bicarbonate buffer pH 8 and reduced for 1 h at room temperature. Iodoacetamide (Sigma) was added to a final concentration of 20 mM, and samples were alkylated for 30 min at room temperature in the dark. Samples were again dried down in the speed-vac, and then digestion was performed as previously described44,45. Briefly, samples were resuspended in 5 mM ammonium acetate pH 4 and digested with endoproteinase GluC at a ratio of 1:20 (enzyme:protein) overnight at room temperature. Digested samples were dried down and then desalted using in-house stage tips44,45. Stage tips were generated by wedging a 0.5 cm circular punch of a 3 M Empore C18 paper disk into the bottom of a P200 pipette tip, followed by addition of porous graphitic carbon (PGC) (HyperCarb, Thermo Fisher Scientific) to a volume three times that of the C18 punch. The stage tips were conditioned with acetonitrile (ACN), equilibrated with 0.1% trifluoroacetic acid (TFA), followed by sample loading in 0.1% TFA, then washed with 0.1% TFA, and eluted with 0.1% TFA in 70% ACN. Two biologic replicates were analyzed for each of the H3WT control and H3R26C samples.
Middle down histone mass spectrometry analysis
The histone samples were analyzed by nanoLC-MS/MS with a Dionex-nanoLC coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). The column was packed in-house using a 75 µm ID × 17 cm column with PGC HyperCarb (3 µm; Thermo Fisher Scientific). The HPLC buffers were A = 0.1% formic acid; B = 80% acetonitrile, 0.1% formic acid. The HPLC gradient was as follows: 2% solvent B for 25 min, then 2% to 13% solvent B in 2 min, from 13% to 16% solvent B in 45 min, 16% to 23% solvent B in 45 min, up to 95% B in 1 min, 95% B for 3 min, and back down from 95% B to 2% B in 1 min and a hold at 2% B for 9 min. The flow rate was at 500 nL/min. Data were acquired using a data-dependent acquisition method, consisting of a full scan MS spectrum (m/z 350–900) performed in the Orbitrap at 120,000 resolution with an AGC target value of 5e5 (normalized AGC target = 125%), followed by selection of peptides of charge states 7–12 and MS2 fragmentation using ETD with calibrated charge-dependent parameters. Dynamic exclusion was set to 10 seconds. Fragmented peptides were detected in the Orbitrap at 30,000 resolution. AGC target was set at 7.5e4 (normalized AGC target = 150%), and maximum inject time set at 200 ms. Histone samples were resuspended in 10 µl buffer A and 4 µl was injected.
Middle down mass spectrometry data analysis
Middle down MS data analysis was performed as previously described44,45. Briefly, raw files were processed in Proteome Discoverer 2.2 (Thermo Fisher Scientific) using the Mascot search engine with a precursor mass tolerance of 2.1 Da and a fragment mass tolerance of 0.01 Da. Carbamidomethyl on cysteine was set as a fixed modification, and variable modifications included acetylation on n-termini and lysine, mono- and di- methylation on lysine and arginine, and trimethylation on lysine. Mascot output files were analyzed with in-house software, HistoneCoderTool ProteoformQuant and IsoScale46,47 (http://middle-down.github.io/Software/), which removes peptides that do not have sufficient fragment ions to map PTMs, and only retains unambiguous peptides with high confidence identification and quantification of PTMs. The output file contains the quantified relative abundance for every identified peptide species, as well as the MS2 level evidence for PTM identification and localization.
Synthesis of Fmoc-Lys(me3)-OH-TFA
Fmoc-Lys(Boc)-OH (6 g) was deprotected by addition of 1:1 trifluoroacetic acid:dichloromethane with stirring for 1 h at room temperature. The solution was concentrated in vacuo overnight and the resultant residue resuspended in 4:1 dichloromethane:MeOH followed by slow addition of N,N-diisopropylethylamine (15 eq.) and then methyl iodide (15 eq.). Reaction progress was monitored by RP-HPLC and quenched by addition of water after 70 min. The product was subjected to a series of extractions with dichloromethane and the final aqueous layer acidified with trifluoroacetic acid to pH 2 and purified by preparatory HPLC. The final product was obtained in 51% yield and characterized by RP-HPLC, ESI-MS, and 1H NMR spectroscopy.
Synthesis of wild-type and arginine mutant H3K27me2 and H3K27me3 histones
Wild-type H2A, H2B, and H4 histones and truncated histone H3.129-135(A29C, C96A, C110A) were recombinantly expressed and purified from E. coli. Peptide hydrazides for semisynthesis were prepared by solid-phase peptide synthesis as previously described48 with Fmoc-Lys(alloc)-OH incorporated at the desired locations for Lys(me2). For synthesis of H3K27me2-containing constructs, peptides were deprotected on resin with Pd(PPh3)4 (0.25 eq.) and N,N-dimethylbarbituric acid (10 eq.) in dichloromethane with agitation (2 × 30 min) and then washed with N,N-diethyldithiocarbamate in dimethylformamide. Resin was subsequently washed with methanol (MeOH) and equilibrated in a 1:1 mixture of PBS:MeOH. Reductive alkylation was performed by addition of 37% (w/w) formaldehyde (50 eq.) in 1:1 PBS:MeOH and solid NaCNBH3 (50 eq.) with agitation (2 × 30 min). Samples were then washed with 1:1 PBS:MeOH, MeOH, and dimethylformamide (sequentially) and peptide cleavage performed as previously described48. Peptide hydrazides were dissolved in activation buffer (6 M guanidinium hydrochloride, 0.2 M NaH2PO4, pH 3.0) and reacted with a solution of NaNO2 (10 eq.) in a salt/ice slurry at −20 °C for 20 min. 2-Mercaptoethanesulfonic acid sodium salt (100 eq.) was then added and the solution pH adjusted slowly to pH 7, after which the reaction was allowed to proceed at room temperature for 15–20 min. The resultant thioesters were purified by semipreparatory RP-HPLC as previously described48. Peptide sequences for histone semisynthesis are shown below:
H3(1-28)K27me2: ARTKQTARKSTGGKAPRKQLATKAARK(me2)S
H3R2A(1-28)K27me2: AATKQTARKSTGGKAPRKQLATKAARK(me2)S
H3R8A(1-28)K27me2: ARTKQTAAKSTGGKAPRKQLATKAARK(me2)S
H3R17(1-28)K27me2: ARTKQTARKSTGGKAPAKQLATKAARK(me2)S
H3R26A(1-28)K27me2: ARTKQTARKSTGGKAPRKQLATKAAAK(me2)S
H3(1-28)K27me3: ARTKQTARKSTGGKAPRKQLATKAARK(me3)S
H3R26A(1-28)K27me3: ARTKQTARKSTGGKAPRKQLATKAAAK(me3)S
For native chemical ligation, co-lyophilized purified peptide thioesters (2 mg) and truncated histone H3.129-135 (2 mg) were resuspended in ligation buffer (6 M guanidinium hydrochloride, 100 mM Na2HPO4, 10 mM TCEP, 10 mM N-acetyl methionine, pH 7.5) with 5% (v/v) 2,2,2-trifluoroethanethiol (TFET). The pH of the solution was carefully adjusted to pH 7.5 and incubated at 37 °C for 4 h, with reaction progress monitored by RP-HPLC. Upon completion, the ligation reaction was diluted two-fold by addition of desulfurization buffer (400 mM TCEP and 40 mM reduced glutathione), degassed, and carefully pH adjusted to pH 7.0. Radical initiator VA-044 was added from a 20x aqueous stock to give a final solution concentration of 10 mM and the reaction allowed to proceed overnight at 37 °C. Reaction progress was monitored and products purified by semipreparatory RP-HPLC. All final histones were characterized by C18 RP-HPLC and ESI-MS.
Preparation of nucleosome arrays
12 x 601 DNA (DNA containing 12 repeats of the 147-bp 601 sequence with 30-bp linkers) was purified from DH5α E. coli as previously described49 with an additional prep cell electrophoresis purification step (Model 491 Prep Cell, Bio-Rad).
601 DNA sequence: ACAGGATGTATATATCTGACACGTGCCTGGAGACTAGGGAGTAATCCCCTTGGCGGTTAAAACGCGGGGGACAGCGCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTCTCCAG
MMTV DNA was prepared by an analogous method, with precipitation by addition of 7.5% (v/v) PEG-6000 used for purification (resultant 155-bp MMTV DNA fragment remains in the supernatant).
MMTV DNA sequence:
ACTTGCAACAGTCCTAACATTCACCTCTTGTGTGTTTGTGTCTGTTCGCCATCCCGTCTCCGCTCGTCACTTATCCTTCACTTTCCAGAGGGTCCCCCCGCAGACCCCGGCGACCCTGGTCGGCCGACTGCGGC ACAGTTTTTTG
Histone octamers were assembled and FPLC-purified as previously described21. 12-mer nucleosome arrays were assembled as previously detailed48 with minor modifications, namely that arrays were precipitated with 4 mM MgCl2 and resuspended array pellets were dialyzed against array assembly end buffer (200 mL × 1 h at 4 °C) to remove MgCl2 from the final array preparations. Arrays were quantified by UV spectroscopy at 260 nm and assembly assessed by agarose polyacrylamide gel electrophoresis with ethidium bromide staining.
PRC2 activity assay
PRC2 core complex (PRC2), consisting of subunits EZH2, EED, RBBP4, and SUZ12, was expressed and purified from Sf9 cells as previously described48. PRC2 methyltransferase activity was assessed by radiometric assay in which 12-mer nucleosome arrays (480 nM 601 sites) were incubated with 20 nM PRC2 in 10 µL histone methyltransferase assay buffer (50 mM HEPES, pH 8.0, 35 mM NaCl, 0.5 mM MgCl2, 0.1% (v/v) Tween-20, 5 mM DTT, 1 mM PMSF, and 0.66 µM [3H]-S-adenosylmethionine) for 1 h at 30 °C. Reactions were quenched by spotting onto Whatman P81 phosphocellulose filter paper (Sigma). Filter papers were air dried for 45 min and then washed 3 × 15 min with 0.2 M NaHCO3 (pH 9.0) with shaking. Filter papers were subsequently dried at 40 °C for 1 h using a gel dryer, submerged in 1 mL Ultima Gold scintillation cocktail, and incubated overnight with shaking. Scintillation counting was performed the following day on a MicroBeta scintillation counter (PerkinElmer), with experimental sample counts corrected for background using a reaction in the absence of enzyme.
CUT&RUN assay
C3H10T1/2 cells were collected by incubation with TrypLE reagent (Gibco) for 2 min at 37 °C followed by quenching with culture media and centrifugation. Cell pellets were resuspended in 1 mL PBS and transferred to a 2 mL screw cap tube. The CUT&RUN protocol was performed in PCR-tubes per the CUTANA (Epicypher) v1.6 protocol except where otherwise noted. In brief, cells were washed twice in wash buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM Spermidine, EDTA-free Protease Inhibitor) prior to resuspending wash buffer. For each CUT&RUN reaction, 500,000 cells were immobilized to Concanavalin-A beads and incubated overnight at 4 °C with antibody diluted 1:100 in antibody dilution buffer (0.005% Digitonin, 2 mM EDTA, Wash Buffer). pAG-MNase (Epicypher, 15–116) digestion was performed for 2 h at 4 °C following which CUT&RUN enriched DNA was purified using phenol-chloroform extraction and ethanol precipitation. NEBNext Ultra II DNA Library Prep kit (NEB, E7645S) was used to prepare sequencing libraries with CUT&RUN enriched DNA. Libraries were sequenced using the Illumina NextSeq 550. Antibodies used were H3 (Abcam, 1791, HA (Biolegend, 901501), H3K4me3 (Active Motif 39159), H3K27me3 (Cell Signaling, 9733), and rabbit anti-mouse IgG (Abcam, 46450).
CUT&RUN analysis
Sequence and transcript coordinates for mouse mm10 and gene models were retrieved from the Bioconductor Bsgenome.Mmusculus.UCSC.mm10 (version 1.4.0) and TxDb.Mmusculus.UCSC.mm10.knownGene (version 3.4.0) Bioconductor libraries respectively.
For the analysis of CUT&RUN data, reads were mapped using the Rsubread package’s align function (version 1.30.6)50. Peak calls made with SEACR (version 1.3, stringent, norm 0.01)51. Consensus peaks were determined to be peaks that were found in the majority of replicates in at least one mutant or control. Peaks were annotated and genome distribution was determined using the ChIPseeker package (Version 1.28.3)52. Enrichment analysis was performed using clusterProfiler (Version 4.0.2)53 against all gene sets from msigdbr (Version 7.4.1) that were related to polycomb or H3K27me3. Peaks were annotated to genes based on proximity; peaks that overlapped with TSS regions were considered for comparisons. Pairwise comparisons were made between the control and mutant using DEseq2 using counts from consensus peaks with significant genes considered as (padj <0.001). Normalized, fragment-extended signal bigWigs were created using the rtracklayer package (Version 1.40.6), and then visualized and exported from IGV.
Ranged heatmaps were generated with profileplyr (Version 1.12.0). Volcano plots were drawn with EnhancedVolcano (Version 1.10.0).
Motif analysis was completed using the MEME suite (Version 5.4.1)54 and its wrapper for R, memes (Version 1.04). Motif enrichment was tested using AME from MEME within the promoter regions of genes that lose H3K27me3 (−1/+0.2 Kb). All promoter regions were used as a background for this test. The CIS-BP database was the source of known motifs (version 2.0).
Bulk RNA-seq and analysis
C3H10T1/2 cells from three separate cultures on different days were collected by washing with PBS, incubation with TypLE for 10 min at 37 °C followed by quenching with culture media and centrifugation at 300 × g. The pellet was resuspended in ice cold PBS, collected at 300 × g at 4 °C, followed by aspiration and flash freezing in liquid nitrogen followed by storage at −80 °C. RNA was then extracted using the RNAeasy kit (Qiagen) with on column DNAase I digestion per manufacturer instructions. From each sample, 500 ng of RNA was used to generate Illumina sequencing libraries using the NEBNext Ultra II RNA Library Preparation Kit (New England Biolabs) with polyA selection according to the manufacturer instructions. Libraries were pooled and single end sequencing (75 bp) was performed using an Illumina NextSeq 500.
Transcript abundance was determined using Salmon (v0.8.1) and the GENCODE reference transcript sequences55. Transcript counts from Salmon were imported into R with the tximport R Bioconductor package (v1.20), and differentially expressed genes were determined with the DESeq2 R Bioconductor package (v1.32)56,57. Read counts were normalized using the rlog function from DESeq2 and z-scores for the indicated gene sets were visualized using the ComplexHeatmap R Bioconductor package (v2.8)58. For GSEA analysis, genes were ranked using the Wald statistic as calculated by DESeq2 and then compared against the indicated gene lists using the clusterProfiler R Bioconductor package (v4.0.5)59,60. Hallmark gene lists were obtained from the Molecular Signatures Database using the msigdbr R package (v7.5.1) and GSEA results were visualized using the enrichPlot Bioconductor R package (v1.12.3)60,61. Gene Ontology analysis was performed using TopGO (v2.40.0) and visualized with Python (see data availability section for access to code).
For the analysis of RNAseq data used in Fig. 4C, transcript expressions were calculated using the Salmon quantification software (version 0.8.2)55. Normalization and differential gene expression analysis were performed using DESeq2 with significant genes considered as (padj < 0.05) (version 1.20.0)62.
Mesenchymal differentiation assay
C3H10T1/2 cells expressing H3WT or H3R26C were cultured in Dulbecco’s Modified Eagle Medium (Corning) with 10% fetal bovine serum, 2 μg/ml puromycin, and 1% penicillin-streptomycin. For the 5-Azacytidine differentiation assay, cells were seeded in 2-well chamber slides (Ibidi, 80286) with 190 cells per chamber. Cells were treated with 3 μM 5-Azacytidine (Sigma A2385) for 24 h following which media was replaced twice a week. After 4 weeks, cells were processed for immunofluorescence. The results from the 5-Azacytidine differentiation assay were analyzed across 4 biological replicates.
Differentiation assay immunofluorescence staining and image capture
Cells undergoing differentiation were fixed with 4% Formaldehyde, washed in PBS and permeabilized (0.1% Triton X-100 for Myosin 4, 0.2% Digitonin for LipidTOX staining). Following permeabilization, cells were blocked with 3% BSA in PBS and incubated overnight at 4 °C with Myosin 4 antibody (Invitrogen14650382, 1 μg/ml) in antibody dilution buffer (2% BSA in PBS), or alternatively incubated for 1 h at RT with LipidTOX (Invitrogen, H34475, 1:200). After overnight incubation with the Myosin 4 antibody, cells were washed in PBS and incubated for 1 h at RT with anti-mouse Alexa Fluor 568 secondary antibody (Invitrogen, A-11004, 2 μg/ml). Cells were washed in PBS and stained with DAPI (Sigma D9542, 2 ug/ml) for 5 min. Samples were rinsed with PBS and Vectashield antifade mounting media (Vector Laboratories, H-1000) was added to the chamber slides. Fluorescence images were taken by imaging the entire slide using Zeiss Celldiscoverer 7 (CD7) automated widefield system with DAPI (Excitation/Emission 353/465 nm), AF488 (Excitation/Emission 493/517 nm), and AF568 (Excitation/Emission 577/603 nm) channels. ZEN 2.6 (Blue edition) software was used for image acquisition.
Differentiation assay immunofluorescence image analysis
ZEN 2.6 (blue edition) was used to stitch tiles and create maximum intensity projections from the original.czi Z-stacks. For quantifying adipogenic differentiation, images with LipidTOX staining were analyzed. For computational efficiency, the entire image was divided into four non-overlapping sub-images. For each sub-image (≈12,000 × 10,002 microns), the area covered by LipidTOX droplets was calculated using the Bernsen Method of Auto Local Threshold (Param1 = 55, Param2 = 0) command in Fiji (version 2.3.0/1.53). The total number of nuclei in each sub-image was calculated using a custom StarDist script in QuPath (version 0.3.0). The total area covered by LipidTOX droplets in the entire image was calculated as the sum of areas covered by LipidTOX in each sub-image. Similarly, the total number of nuclei in the entire image was calculated as the sum of the number of nuclei in each sub image. Adipogenesis quantification was adapted from established methods35 and was quantified as the Adipogenic Index (AI), which is expressed as the percentage area covered by LipidTOX normalized to the total number of nuclei for each sample (arbitrary units) (Supplementary Fig. S8). Average values of AIs across 4 biological replicates were plotted and ratio paired t-test was performed for statistical analysis.
For quantifying myogenic differentiation, four non-overlapping regions (≈7955 × 4290 microns) with Myosin 4 signal were analyzed for each sample. These regions were analyzed in QuPath software (version 0.3.0). Myosin 4 positive cells were outlined manually by using the annotation tool in QuPath and number of nuclei in each Myosin 4 positive cell annotation was quantified using a custom StarDist script. The total number of Myosin 4 positive cells and the number of Myosin 4 positive cells with more than one nuclei was used to calculate the percentage of multinucleated Myosin 4 positive cells. Average values of the percentage of multinucleated Myosin 4 positive cells across 4 biological replicates were plotted and a Mann–Whitney test was performed for statistical analysis.
Histone mutant immunofluorescence and imaging analyses
HEK293T cells stably expressing HA-tagged wild type H3.1 and a panel of arginine mutants were grown in 12-well plates with 18 mm coverslip placed on the bottom (Neuvitro GG-18-1.5). Upon reaching 50–75% confluence, cells were rinsed in PBS, fixed with 1% paraformaldehyde in 0.1% Triton X-100 in PBS (PBS-T) for 20 min, washed with PBS-T and blocked for one hour in 1% BSA in PBS-T. Coverslips were then incubated with primary antibodies to HA epitope (mouse HA.11, BioLegend 901503, 1:200) and total H3 (rabbit ab1791, abcam, 1:1000) in 1% BSA-PBS-T solution in humidity chambers overnight, washed three times with PBS-T, and incubated with AlexaFluor-conjugated secondary antibodies (Invitrogen donkey anti-rabbit AF488 (A21206) and goat anti-mouse AF568 (A11031)) diluted 1:1000 in 1% BSA-PBS-T for four hours. Coverslips were then washed twice with PBS-T, incubated for 10 min with DAPI (1 µg/ml final concentration) in PBS, rinsed with PBS, and mounted on glass slides with ProLong Gold reagent (Invitrogen P36934). Slides were imaged using Zeiss LSM 780 AxioObserver, C-Apochromat 40x/1.2 water objective, and Zen suite (Rockefeller University BioImaging Resource Center). Single confocal slices were minimally processed in ImageJ and assembled in Adobe Illustrator CC.
Single cell RNA-seq and analysis
C3H10T1/2 cells expressing H3WT or H3R26C were differentiated using 5-Azacytidine as described above. On day 28, cells were washed with PBS and dissociated using TrypLE Express, which was quenched with Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum. The cells were collected by centrifugation and resuspended in 0.04% BSA in PBS. The cells were passed through a 70 µm cell strainer and 50,000 cells were submitted to the Memorial Sloan Kettering Integrative Genomics Operation core facility for further processing and sequencing on a 10X genomics platform.
For analysis, the FASTQ files were first processed using CellRanger V6 with the mouse genome and transcript reference data (refdata-gex-mm10-2020-A) supplied by 10X genomics. The output from CellRanger was then analyzed using the Seurat R package version 4. The filtered barcode/feature matrix was read using Seurat read10X command and the percentage of mitochondrial reads per cell was computed for subsequence filtering. Cells were filtered using the following criteria: percent mitochondrial read greater than 20% or the number of genes was less than 1500 or the number of distinct molecules was less than 5000. If any of these conditions were true the cell was filtered out of the downstream analysis. Next, we scored the cell cycle phase of each cell using Seurat’s scoreCellCycle functions. The list of cell cycle genes for mouse was determined by taking the supplied list of human cell cycle genes and mapping them to mouse genes using the bioMart R package to do the homology mapping.
The filtered data was normalized and scaled using the SCTransform method from Seurat. For the transform we regressed against the cell cycle scores previously computed. After normalization we computed the PCA coordinates and retained the first 20 coordinates in the clustering and projection analysis. For clustering we used the Seurat FindNeighbor and FindClusters functions with several resolution values and after manual inspection fixed on a resolution value of 0.2 for subsequent work. We also computed the UMAP project using RunUMAP and 20 pca coordinates. Cluster specific marker genes were computed using FindAllMarkers with a cutoff of 0.25 in the log fold change and a minimum percentage of 25%. For two specified gene sets: Adipocyte63 and Skeletal Muscle [https://www.gsea-msigdb.org/gsea/msigdb/cards/SKELETAL_MUSCLE_DEVELOPMENT] (Supplementary Data 10), we computed a score for each using AddModuleScore.
Trajectory analysis was done using Monocle3 (version 1.3.1). We used the R package SeuratWrappers to convert the Seurat objects for use in Monocle preserving the original pca mapping and UMAP reduction. The data was re-clustered with Monocle’s cluster_cells function and then the trajectory graph and cell pseudotimes were computed.
All of the custom R scripts used in this analysis are available as described in the code availability section. The following software packages were also used: CellRanger Version 6 (https://support.10xgenomics.com), R Version 4.1.2 (https://www.r-project.org), Seurat Version 4.2.0 (https://satijalab.org/seurat), Monocle3 Version 1.3.1 (https://cole-trapnell-lab.github.io/monocle3), SeuratWrappers Version 0.3.1, and SeuratObject Version 4.1.3.
Teratoma formation and histologic analysis
To generate teratomas, 1 × 106 mESC expressing H3WT or H3R26C were injected subcutaneously in each flank of 6–7 weeks old female NOD.Cg-Prkdcscid immunodeficient mice (Jackson Laboratories, Strain 005557). The mice were housed in ambient temperatures from 68-79 degrees Fahrenheit, 30–70% humidity, with light/dark times of 0700/1900 hours. For subcutaneous injections, mESC were mixed 1:1 with Matrigel Basement Membrane Matrix (Corning, 356231) and a total volume of 200 μL was injected in each flank. After 3 weeks, tumors were excised immediately postmortem and fixed in 10% Neutral Buffered Formalin (v/v). Mice were treated in accordance with a protocol approved by the Rockefeller University Institutional Animal Care and Use Committee. The maximal allowed tumor size/burden was 1.5–2 cm or ulceration; this limit was not exceeded. Fixed tumors were submitted to the Laboratory for Comparative Pathology at the Memorial Sloan Kettering Cancer Center for processing, embedding, sectioning, and hematoxylin and eosin staining. Germ layer proportions were assigned by a board-certified veterinary pathologist (SCS). Sex was not considered in the study design or analysis since the experiments were designed and controlled to test the effect of a histone mutation as the variable of focus.
Statistical & reproducibility
The statistical methods for analysis are described in the relevant contexts in the methods, figure legends, or embedded in published or custom code (see code availability section). Investigators performing histologic analysis of teratomas were blinded to the genotypes of the mESCs used to generate the teratomas. The experiments were not randomized. Sample size is described in the figure legends and/or methods section. No statistical method was used to predetermine the sample size. No data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-024-49486-5