Human tissue samples
Tissue samples and annotated data were obtained, and experimental procedures were performed within the framework of the non-profit foundation HTCR, including informed patient consent.
Intestinal organoid and tumouroid cell culture
Supplementary Table 1 provides anonymized basic donor information. After isolation of intestinal crypts following a previously described protocol27, crypts were embedded in 25 µl droplets of growth factor-reduced Matrigel (356231, Corning) and cultured in a 50% (v/v) mix of Human IntestiCult OGM human basal medium (OGM; 100-0190, StemCell) and organoid supplement (100-019, StemCell Technologies) in a 24-well clear TC-treated plate (3524, Corning Costar). Y27632 (10 µM l−1; 72302, StemCell Technologies) and 1% penicillin/streptomycin (15140-122, Gibco) were spiked into the media after seeding the organoids. Throughout the week of expansion, OGM was changed every 2 d, deprived of Y27632. Organoids were passaged every 7 d, up to a maximum of 25 passages. After 2–3 passages post-isolation, organoid cultures were amenable to experiments. Isolation and passage protocol in ref. 27 were slightly adjusted for our purposes34. Briefly, we added 500 µl of Gentle Cell Dissociation reagent (07174, StemCell) before disrupting the domes. After 15 min of incubation at r.t., the suspension was centrifuged at 250 g for 5 min before washing with ‘organoid wash’ (DMEM-F12 + Glutamax (61870-010, Gibco), 10 mM HEPES buffer (15630-056, Gibco), 1% penicillin/streptomycin, 1% bovine serum albumin (A9647-100G, SIGMA)). Centrifugation was repeated and the supernatant discarded; the pellet was then resuspended and seeded in a Matrigel dome.
Colorectal cancer organoids (termed tumouroids) were obtained and handled as described above, except for media applied. Tumouroids were grown in a 50% (v/v) mix of IntestiCult OGM human basal medium (100-0190, StemCell) and DMEM-F12 + Glutamax, supplemented with 1% penicillin/streptomycin and 10 µM l−1 Y27632. Media change and passages were performed as described for the healthy organoids.
For differentiation of the organoids, OGM was removed after 5 d of expansion. Human IntestiCult ODM human basal medium (ODM; 100-0212, StemCell Technologies; 500 µl, 50% v/v) and organoid supplement (100-019, StemCell Technologies) were added instead and supplemented with 100 µM l−1 N-[N-(3, 5-difluorophenacetyl)-l-alanyl]-S-phenylglycinet-butyl ester (DAPT) (72082, StemCell Technologies). Differentiation media were changed every 2 d until usage for co-culture assay or fixation of the organoids.
Intestinal organoid–PBMC co-culture
Intestinal organoids and tumouroids were cultivated and differentiated as described earlier. Three days after the initiation of differentiation, organoids and tumouroids were collected. Domes were washed in the plate with 500 µl cold 1X Dulbecco’s phosphate-buffered saline (1X DPBS) (2345154, Gibco), followed by incubation with 500 µl cell recovery solution (35423, Corning) at 4 °C for 40 min. Next, organoid and tumouroid domes were disrupted by gentle up- and down-pipetting close to the dome. We then collected and transferred the suspension to a 15 ml Falcon tube, centrifuged it for 5 min at 200 g, followed by a wash with ‘organoid wash’ (see above for composition) and repeated centrifugation. Organoids and tumouroids were kept on ice until resuspension with PBMCs.
PBMCs were thawed following a standard thawing protocol. Briefly, PBMCs were thawed for 2 min in a 37 °C water bath. The cells were then transferred to a 15 ml Falcon tube. The PBMC vials were carefully rinsed with pre-heated 37 °C PBMC media (RPMI 1640 + Glutamax, 10% fetal bovine serum (heat inactivated) (97068-085, VWR), 1% penicillin/streptomycin (100X, 11140-035, Gibco), 1% non-essential amino acids (11360-039, Gibco, 100X), 1% sodium pyruvate (100X, 15140-122, Gibco), plus 1:100 DNAse (DNAse I, 0453628001, Roche)). The suspension from the rinsed vial was added drop by drop to the cells in the 15 ml Falcon tube. Next, a further 6 ml of PBMC medium including 1:100 DNAse was added to the cells before centrifugation at 320 g for 5 min. The supernatant was discarded before the cell pellet was resuspended in PBMC medium to yield ~4 million cells per ml. The resuspended cells were transferred to the incubator (37 °C, 5% CO2) to rest for 15 min. Next, PBMCs were counted with Trypan Blue (216040, Invitrogen) on a Countess II (Invitrogen). After centrifugation at 320 g for 5 min, PBMCs were then resuspended in PBMC medium at 4 million cells per ml and transferred in a 50 ml tissue culture flask (353014, Corning). The flask was placed slightly tilted in the incubator for a minimum of 4 h or overnight. After the incubation, PBMCs were transferred to a Falcon tube, centrifuged and stored on ice until resuspension with the intestinal organoids. Depending on the application, different matrices were generated.
In general, the number of PBMCs needed was calculated using the following formula: PBMCs needed = (V (µl)×22,000)*n, where V is the volume of dome and n is the sample size. Immune-cell number was guided by the findings of Fu et al.52 Dome volumes varied between imaging methods:
-
1.
Killing assay: organoid–PBMC co-culture in a 100% Matrigel matrix for live imaging and fixed whole-mount imaging
Domes of 10 µl per sample (V) were prepared to be seeded in a μ-Plate Angiogenesis 96-well black plate (69646, ibidi). First, PBMCs were resuspended in the necessary volume of 100% Matrigel. Second, the PBMC-matrix suspension was carefully mixed with the organoid pellet to decrease the likelihood of organoid damage. To avoid phase separation and Matrigel polymerization during the seeding process, the suspension was kept in a trough on ice. If multiple organoid lines were seeded simultaneously, organoid–PBMC suspensions were kept in PCR strips on ice to ease the seeding process, keeping the suspension cold, thus allowing continuous resuspension and faster handling using a multichannel pipette. After seeding the co-culture blend, the plates were flipped carefully. The flipped plate was placed in the incubator for 15 min. During the polymerization of the domes, PBMC media and ODM were prepared (50% v/v). Y27632 (10 mMol l−1) was spiked into the blend. Media were added to the wells.
-
2.
Organoid–PBMC co-culture in a 50% (v/v) collagen–Matrigel matrix for histology-based mIF imaging
For the co-culture setup ultimately embedded in FFPE blocks, 25 µl domes (V) of Matrigel and collagen (50% v/v) were prepared and seeded in a 24-well clear TC-treated plate. For the creation of the collagen–Matrigel matrix, 3D Culture Matrix rat collagen I (3447-020-01, R&D systems) was used. The collagen I was prepared by first using 1 M HEPES (15630-056, Gibco), followed by 37 mg ml−1 NaMCO3 (150 mM NaCl, 1 M HEPES) and finally collagen I at a ratio of 1:1:8. Matrigel and the collagen I mixture (50% v/v) were then mixed thoroughly. PBMC and organoid or tumouroid resuspension, dome polymerization and media preparation were executed as described in the previous paragraph.
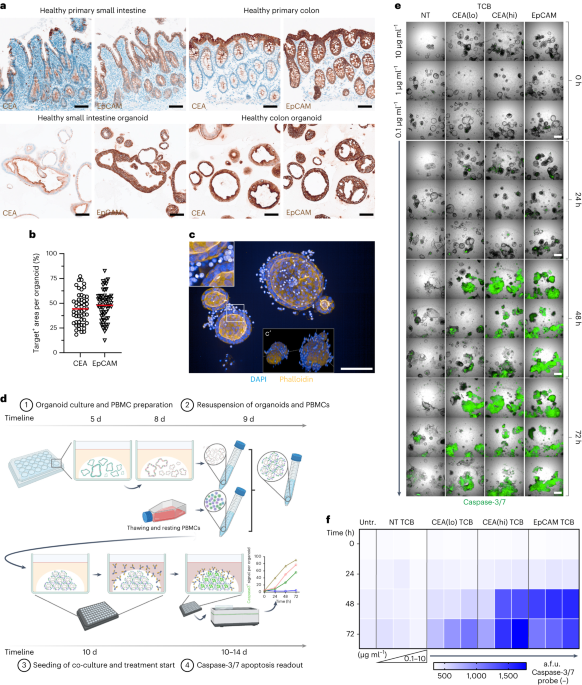
Approximately 40 organoids per well was determined to yield the best results for the killing assay, both in terms of organoid segmentation during image analysis as well as statistical power, whereas ~100 organoids were seeded when performing the FFPE-based imaging approach in 25 µl domes. Exact organoid to PBMC cell number was confirmed by mIF quantification across dozens of samples at baseline (0 h condition). Analysis of the images yielded a 1.1:1 E:T ratio (considering EpCAM− cells as the entire PBMC population). Only considering the main effectors (CD4+ and CD8+ T cells) resulted in a 1:2.5 E:T ratio. Given the 10 µl dome for the killing assay, for example, this results in 220,000 PBMCs to ~200,000 epithelial cells, or 550,000 PBMCs to 500,000 epithelial cells in a 25 µl dome.
T-cell bispecific antibodies for treatment
We previously provided detailed information about the construction of the CEA TCBs and NT TCB53. The trivalent 2 + 1 IgG EpCAM TCB was generated with the ‘Knob-into-holes’ technique, resulting in a monovalent CD3+ lymphocyte arm as well as a bivalent target (EpCAM) engaging arm. All TCBs plus the corresponding NT TCB were titrated in PBMC–organoid media and applied 24 h post co-culture seeding, which denotes treatment day 0. Media were not changed after initiation of treatment.
Blocking experiments
Where indicated, recombinant human monoclonal antibody etrolizumab (MA5-41929, Invitrogen; 10 µg ml−1) and anti-integrin β7 monoclonal antibody (I-1141, Leinco; 10 µg ml−1) were applied together with TCBs and re-titrated daily without changing the initial TCB-containing media.
Low-resolution killing assay
Apoptosis was assessed using the CellEvent Caspase-3/7 detection reagent (C10423, Invitrogen) during the TCB-treatment period. CellEvent Caspase-3/7 detection reagent was diluted 1:1,000 and added to the respective master mix. Samples were imaged in confocal mode at ×5 magnification (air objective) with the Operetta CLS high-content imaging device (Perkin Elmer) covering a ~450 µm Ζ-stack, starting at −150 µm. The distance between Ζ-stacks was set to the minimum of 27 µm for the ×5 objective (autofocus: two peak; binning: 2). Channels selected were brightfield and the predefined Alexa 488. Per well, 5 fields were acquired, covering nearly the entire surface of the 96-well PhenoPlate. Given these imaging parameters, an imaging run of an entire 96-well plate took 2 h and 39 min. To rule out time-related killing impacts of TCBs applied between different plates, the time difference was accounted for by delaying TCB application per plate. The 72-h time course was imaged live every 24 h, starting with the 0 h baseline. CO2 was set to 5%, temperature to 37 °C. Caspase-3/7 fluorescence signal intensity was quantified using the Opera Harmony software (Perkin Elmer). Briefly, segmentation of organoids was done by using ‘Find Texture Regions’ on the basis of the brightfield signal only, followed by ‘Select Region’ and ‘Find Image Region’ to segment single organoids as objects. Next, ‘Calculate Morphology Parameters’ was performed to select objects >7,500 µm2 with ‘Select Population’. Next, the caspase-3/7 fluorescence signal per individual organoid was determined using ‘Calculate Intensity Properties’ of the AF 488 channel within these objects.
Flow cytometry
Five hours before collection, cultures were treated with eBioscience Protein Transport Inhibitor Cocktail (500X, ThermoFisher) to facilitate intracellular accumulation of temporally expressed soluble proteins. Duplicate wells of PBMC–organoid co-cultures from each condition were collected at 5, 24, 48 and 72 h post treatment. Co-cultures were first washed with PBS and then digested to single cells using Accutase solution. Cell suspensions were passed through a 70 µm strainer and stained for surface proteins. Cells were then fixed and permeabilized using the eBioscience Foxp3 Transcription Factor Staining Buffer set (ThermoFisher) and subsequently stained for intracellular and intranuclear proteins (Supplementary Table 2). Stained cell suspensions were acquired on a BD Fortessa X-20 flow cytometer (BD Biosciences) and analysed using FlowJo v.10.
Analysis of cytokines
At each timepoint (0, 24, 48, 72 h) after TCB administration, 50 µl of supernatant was collected from the 24-well clear TC-treated plate. Supernatants were quickly centrifuged to remove cell debris and then frozen at −20 °C until further processing. Measurement of the cytokines (GM-CSF, IFNγ, TNFɑ, IL-2, IL-4, IL-6, IL-8, IL-10) was performed using the BioPlex Pro Human Cytokine 8-plex assay kit (M50000007A, Bio-Rad). The controls, standards and beads were prepared by following manufacturer instructions. Briefly, we diluted the sample 1:2 according to guidelines. After preparing the 4-fold dilution series, we vortexed the beads and added them to the provided assay plate. After washing the plates with a plate washer (405TS, Bioteck), standards were added and incubated for 30 min at r.t. on a rocker at 850 r.p.m. (±50) as recommended. After another wash, detection antibodies were added, followed by incubation for 30 min at 850 r.p.m. before washing again. Lastly, we added the streptavidin-phycoerythrin mix, incubated the samples for 10 min at 850 r.p.m., ending with washing and resuspension of the beads with the provided assay buffer. The Luminex assay was performed with a BioPlex 200 instrument (Bio-Rad). First, the standard and control plates were measured, yielding a standard curve for each cytokine. Second, a quick validation of the concentration ranges confirmed the kit to be useful. Third, plates of interest were measured. With the previously generated best curve fit of the standards, the mean fluorescence intensity (MFI) of the samples was automatically calculated to the concentrations in pg ml−1.
FFPE-embedding of co-cultures
To FFPE-embed the co-cultures, the samples were seeded in a 50% (v/v) Matrigel–collagen I matrix as explained above. Wells were washed once with 1X DPBS before fixation with 4% paraformaldehyde (PFA) in the 24-well clear TC-treated plate. After 30 min of fixation at r.t., the wells were washed three more times before complete aspiration of the 1X DPBS. In the meantime, HistoGel (22-110-678, Thermo Scientific) was heated up according to manufacturer instructions. Pre-liquefied HistoGel (400 µl) was dispensed into the 24-well clear TC-treated plates. The plates were then placed in a 4 °C refrigerator for ~10 min. After polymerization of the HistoGel, the organoid–HistoGel ‘platelet’ was carefully lifted out of the 24-well clear TC-treated plate using a thin metallic spatula. If parts of the sample adhered to the plate, the collagen–Matrigel matrix was not of satisfactory quality. The samples were then distributed into biopsy cassettes and dehydrated overnight using a vacuum filter processor (Sakura, TissueTek VIP5). On the next day, the samples were embedded in liquid paraffin in metallic moulds and capped with the corresponding FFPE-block cassette. After polymerization of the paraffin, metallic moulds were removed and FFPE blocks stored at −20 °C until sectioning.
Microtome sectioning
As the polymer structure of the embedded HistoGel differs substantially from that of the paraffin, organoid detection within the HistoGel was easily possible during sectioning. FFPE blocks were sectioned at a thickness of 3.5 µm and transferred on Superfrost Plus Adhesion microscope slides (J1800AMNZ, Epredia). Slides were incubated in a slide oven overnight at 37 °C, with the specimen facing up to prevent potential loss of organoids due to melting paraffin.
H&E staining
H&E staining was executed in a fully automated manner following the standard protocol on the Ventana HE600 stainer (Roche Tissue Diagnostics).
IHC staining
IHC staining of FFPE slides was performed using Ventana Discovery Ultra automated tissue stainer (Roche Tissue Diagnostics). Slides were baked first at 60 °C for 8 min and subsequently further heated up to 69 °C for 8 min for subsequent deparaffinization. This cycle was repeated three times. Heat-induced antigen retrieval was performed with Tris-EDTA buffer (pH 7.8; CC1, 950-227, Ventana) at 95 °C for 32 min. After blocking with Discovery Inhibitor (760-4840, Ventana) for 8 min, normal goat serum at 2% or 10% was applied as pre-treatment. Afterwards, primary antibodies (Supplementary Table 3) were diluted in Discovery Ab diluent (760-108, Ventana) and applied in the concentrations determined in previous establishment runs. Primary antibodies were detected using anti-species secondary antibodies conjugated to horseradish peroxidase (HRP) (OmniMap anti-rabbit HRP, 760-4311; OmniMap anti-mouse, 760-4310, Ventana; OmniMap anti-rat, 760-4457, Ventana) for 16 min and subsequently visualized by conversion of 3,3’-diaminobenzidine (DAB) (Discovery ChromoMap DAB kit, 760-159, Ventana) or Discovery. Lastly, specimens were counterstained with haematoxylin (Haematoxylin II, 790-2208, Ventana) and bluing reagent (760-2037, Ventana). After dehydration with a standard series of alcohol (75%, 95%, 100%, 100% v/v; CAS64-17-5, Roche) and Xylol baths (100% v/v, 444240050, ACROS Organics), slides were mounted in a fully automated manner using the RCM7000 cover slipper (MEDITE) and a standard histoglue (00811-EX, Pertex). Slides were dried for at least 2 h before imaging.
Brightfield imaging
HE and IHC stained slides were imaged with a brightfield whole-slide scanner at ×40 (Hamamatsu, NanoZoomer S360). Pixel size was 0.23 µm px−1 in all brightfield images.
Brightfield image analysis
Briefly, single organoids were automatically detected using a deep-learning algorithm trained to distinguish matrix and organoids or tumouroids (iterations: 1,030; cross-entropy: 0.08; DenseNet AI V2 Plugin). After quick validation, organoids and tumouroids were detected as objects and labelled as individual regions of interest (ROIs) (Supplementary Fig. 3b–d). Only objects >7,500 µm2 were deemed positive. Area Quantification v.2.3.1 and Area Quantification FL v.2.2.2 modules were used to quantify positive-staining regions against overall size of each individual organoid and tumouroid, respectively (Supplementary Fig. 3b,d). Isotype controls and secondary-only negative controls on the tissue of origin served as a negative signal threshold to prevent biased adjustments.
FFPE-based mIF staining
mIF staining of FFPE slides was performed using Ventana Discovery Ultra automated tissue stainer (Roche Tissue Diagnostics). Slides were first baked at 60 °C for 8 min and subsequently further heated up to 69 °C for 8 min for subsequent deparaffinization. This cycle was repeated twice. Heat-induced antigen retrieval was performed with Tris-EDTA buffer (pH 7.8; CC1, 950-227, Ventana) at 92 °C for 32 min. After each blocking step with Discovery Inhibitor (760-4840, Ventana) for 16 min, the Discovery Inhibitor was neutralized. Primary antibodies were diluted in 1X Plus Automation Amplification diluent (FP1609, Akoya Biosciences). Primaries were detected using anti-species secondary antibodies conjugated to HRP (OmniMap anti-rabbit HRP, 760-4311; OmniMap anti-mouse, 760-4310, Ventana; OmniMap anti-rat, 760-4457, Ventana) (Supplementary Table 3). Subsequently, the corresponding Opal dye (Opal 480, OP-001000; 520, OP-001001; 570, OP-001003; 620, OP-001004; 690, OP-001006; 780, OP-001008; Akoya Biosciences) was applied. After every application of a primary, corresponding secondary antibody and opal dye, an antibody neutralization and denaturation step was applied to remove residual antibodies and HRP, before starting the staining cycle again with the Discovery Inhibitor blocking step. Lastly, samples were counterstained with 4’,6- diamidino-2-phenylindol (DAPI, Roche). Sequential order of the primary antibodies as well as corresponding dyes were determined during establishment runs. Neutralization of HRP and denaturation of the proteins were performed after every primary antibody cycle to avoid cross-bleeding and cross-reacting antibodies.
FFPE-based mIF imaging
mIF stainings using the Opal dyes from Akoya were digitized using multispectral imaging by the Vectra Polaris (Perkin Elmer) employing the MOTiF technology at ×20 magnification for all 7 colours (Opal 480, Opal 520, Opal 570, Opal 620, Opal 690, Opal 780 and DAPI). Laser exposure and intensity settings were adjusted on multiple slides per staining panel. Slides were scanned in a batch manner to ensure the same imaging settings and cross-comparability for later image analysis with HALO AI. Next, unmixing of the channels and tiling of the images were performed with PhenoChart (v.1.0.12) and inForm (v.2.4). Raw image data were saved as .qptiff. Tiles were fused in HALO (Indica Labs, v.3.2.1851.328). Pixel size in these images was 0.50 µm px−1.
FFPE-based mIF image analysis
Image analysis of the IF images was performed with HALO AI (Indica Labs, v.3.2.1851.328). Briefly, single organoids were automatically detected using a deep-learning algorithm trained to distinguish between matrix and organoids or tumouroids, (iterations: 11,415; cross-entropy: 0.428; DenseNet AI V2 Plugin). After quick validation, organoids and tumouroids were detected and labelled as individual ROIs, objects (Fig. 4c). Only objects >7,500 µm2 were considered positive.
Annotation layers and cell segmentation for immune cell quantification
The ROI of the objects allowed generation of one inward and two outward concentric partitions (25 µm margins). To distinguish the four zones for subsequent analysis, the outer concentric outline was drawn as inclusion ROI (solid line), whereas the concentric outline defining the beginning of the next outline had to be copied and inverted as exclusive ROI (dotted line) into the corresponding annotation layer of the previous margin. This resulted in an annotation layer per zone, which allowed quantification of immune cells within (Fig. 3c and Supplementary Fig. 3f). The HighPlex FL v.4.1.3 module was used to perform nuclear segmentation on the basis of DAPI+ cells. For detection and subsequent quantification, DAPI+ cells and distinct markers for each individual cell type of interest were merged. Thus, secondary-only negative controls on the tissue of origin served as a negative signal threshold to prevent biased adjustments on the test slides. The analysis module was deployed on ROIs per single object (organoid or tumouroid). Where indicated, normalization to object area was performed. Overlapping organoid and tumouroid zones were deleted.
Quantification of E-cadherin+, panCK+ and Caspase-3+ epithelium
Quantification of TCB-triggered epithelial killing via increase in caspase-3+ (apoptosis) and decrease in panCK+ epithelium (loss of integrity) of the organoids and tumouroids was performed by harnessing the DenseNet AI V2 classifier trained to distinguish the matrix, organoids, caspase-3+ and the panCK+ signal in the 25 µm margin of epithelium of both organoids and tumouroids (Supplementary Fig. 3e; iterations: 34,035; cross-entropy: 0.5; HALO AI). For each class, the area covered was quantified and calculated against the overall area of epithelium of the objects, yielding distinct percentages.
Whole-mount immunofluorescence staining
Before fixation with 4% PFA (43368, Alfa Aesar) for 30 min at r.t., samples were washed with 1X DPBS (14287-050, Gibco). After fixation, samples were washed three more times with 1X DPBS on a rocker for 15 min at 150 r.p.m. Next, 1X DPBS was removed and samples were stained with DAPI (1:2,000 dilution; 62248, Thermo Scientific), Phalloidin-Atto 594 (1:3,000 dilution; 51927, Sigma) and Caspase-3/7 (1:1,000 dilution; C10423, Invitrogen). After 90 min incubation at r.t., samples were washed three times with 1X DPBS, again using a rocker. Images were acquired with the Operetta CLS (Perkin Elmer) using a ×20 water objective. Optical mode, focus and binning were set up as described above. The first plane imaged was at −15 µm, followed by 50 z-stacks with a distance of 4 µm. 3D reconstruction was performed in Harmony (Perkin Elmer).
Statistics
Details about statistical analysis are provided in each figure’s description. Unpaired t-tests were applied to compare two datasets. One-way and two-way analysis of variance (ANOVA) including Tukey’s multiple comparison tests were used for more than two datasets. Datasets are visualized with mean and standard deviation as indicated in the figure legends, unless documented otherwise. P values to indicate statistical significance are indicated in the figures. ROUT outlier analysis with low Q coefficients in GraphPad Prism 8 (GraphPad Software) was performed to remove definite outliers, as indicated in figure captions. All graphs in the document were generated in GraphPad Prism 8.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41551-023-01156-5