Ethics
This study involved undertaking animal procedures in four different countries: U.K, USA, Austria, and the Republic of South Africa. Animal procedures were carried out in accordance with Home Office, UK regulations and the Animals (Scientific Procedures) Act, 1986 of UK, the Institutional Animal Care and Use Committee (IACUC) of USA, Act 7, 1991 of South Africa, and the Directive 2010/63/EU of the European Parliament.
Normal human colonoscopy samples were collected under the research tissue bank ethics 16/YH/0247 supported by NIHR Biomedical Research Centre, Oxford, U.K. and under the London Dulwich Research Ethics Committee (reference number 15/LO/1998). Written informed consent was obtained from all participants undergoing routine bowel cancer or IBD screening. All samples were anonymized.
Mouse husbandry
Wild-caught mice (F1) were acquired from a founder population trapped in lower Austria and Vienna (2016) and housed at the Konrad Lorenz Institute of Ethology, University of Vienna, Austria. All C57BL/6 J mice used in this study were purchased from Charles River (Kent, UK) or the Jackson Laboratory (USA) and housed at Biomedical Services Unit in John Radcliffe Hospital, Oxford, UK or at Rutgers University Animal Facility in Newark, New Jersey, USA. Mice were housed in individually ventilated cages under specific pathogen-free conditions and maintained at 19–23 °C temperature with 45-65% relative humidity, in an alternating 12-h light/12-h dark cycles and fed with food and water ad libitum.
Naked mole rat husbandry
Naked mole rats (NMRs) were housed at the Animal Facility of the Department of Zoology and Entomology, University of Pretoria. The NMRs were kept in tunnel systems consisting of several Perspex chambers containing wood shavings as nestling material. The NMR room was maintained at temperatures ranging between 29–32 °C, with relative humidity around 40-60%. NMRs were fed chopped fresh fruits and vegetables (apple, sweet potato, cucumber, and capsicum) daily ad libitum along with weekly supplement of ProNutro (Bokomo). Since NMRs obtain all their necessary water from food sources, no drinking water was provided to the animals. All scientific procedures on NMRs were conducted under ethics approval (NAS046-19 and NAS289-2020) by the Animal Ethics Committee, University of Pretoria. In addition, DAFF section 20 approval was granted (SDAH-Epi-20111909592).
For all analyses, both male and female mice, NMRs, and humans were included in the study.
In vivo administration of BrdU and EdU
15 mg/mL solution of BrdU (5-bromo-2’-deoxyuridine, Abcam, ab142567) and 12.3 mg/mL solution of EdU (5-ethynyl-2-deoxyuridine, Merck, 900584) were prepared in sterile 1× PBS (Gibco, 10010023) and filtered through a 0.2 µm strainer. Using a 27-gauge needle and 1 mL syringe, 100 mg per kg bodyweight BrdU and 82.14 mg per kg bodyweight EdU were administered intraperitoneally. Animals were checked regularly for signs of discomfort (hunched back, shivering, low mobility) after the injection.
For cumulative labelling protocol using BrdU, the first injection in naked mole rats was administered between 14:00 to 15:00. Subsequent BrdU injections were given every 8 h for a duration of 5 days and intestinal tissues were collected every 8 h after the first injection. In C57BL/6 J mice, the first BrdU injection was also given between 14:00 to 15:00, with further injections given every 6 h for a total of 2.25 days. Mouse intestinal tissues were collected 1 h after each injection. The rationale for the frequency and total number of injections in the two species is discussed in Supplementary Note 1.
In vivo administration of dextran sulphate sodium
Dextran sulphate sodium (DSS) salt (Merck, 42867) was dissolved in sterile ddH2O to prepare 0 to 8.75% (w/V) solution. Using a 2 mL syringe fitted with a plastic feeding tube (Prime Bioscience, FTP-20-38), 50 mL per kg bodyweight of DSS solution in NMRs or 12 mL per kg bodyweight in mice was administered orally at specific intervals for 3 days. Body mass was monitored daily and stool samples collected while animals were also checked for signs of discomfort (e.g. hunched back, shivering, low mobility) every 3 h.
Intestinal tissue collection and processing
After sacrificing the animals by approved procedures, the intestine was immediately isolated from the abdominal cavity and fatty tissue was removed. The small intestine was then divided into three equal sections: SB1 (duodenum), SB2 (jejunum) and SB3 (ileum). All three parts of the small intestine and colon were then flushed with 1× PBS (Phosphate Buffered Saline) solution using a P1000 pipette to clean all the faecal material. Each tissue section was then cut open longitudinally using a gut cutting device86 and the edges pinned down onto a 3MM filter paper such that the luminal side was facing upward. The tissue was then fixed in 10% neutral buffered formalin overnight at room temperature. The following day fixed intestinal tissues were rolled using the Swiss-rolling technique87 and stored in 70% ethanol at 4 °C. Next, formalin-fixed Swiss-rolls were dehydrated through increasing concentrations of ethanol, cleared through xylene, and embedded in paraffin. The paraffin blocks were sectioned at 4 µm thickness using a microtome (Anglia Scientific).
Haematoxylin and Eosin staining
Tissue sections on SuperFrost Plus slides (VWR, 6310108) were deparaffinized by submerging slides in xylene (2 times, 10 min each) and rehydrated in 100% ethanol (2 times, 5 min each), 95% ethanol (2 min), 70% ethanol (2 min), 50% ethanol (2 min), and distilled water (5 min). Sections were then stained with Harris Haematoxylin (Merck, HHS32) for 2 min 45 s followed by washing in running tap water for 5 min. Next, slides were dipped in 95% ethanol ten times before sections were counter-stained with Eosin solution (Merck, 117081) for 3 min. This was followed by tissue sections being dehydrated in 95% ethanol (15 s) and 100% ethanol (2 times, 15 s each), dipped in xylene (2 times, 5 min each), and finally coverslipped using DPX Mountant (Merck, 06522).
Alcian blue staining
Tissue sections on SuperFrost Plus slides (VWR, 6310108) were first deparaffinized with xylene (2 times, 5 min each). They were rehydrated in 100%, 90%, 70% ethanol (5 min each) and tap water (2 min), dipped in 3% acetic acid solution (3 min) before staining with Alcian blue 8GX (Merck, A5268) solution (pH 2.5) for 30 min. Tissue sections were then washed (5 min) in running tap water and counterstained (5 min) with Nuclear Fast Red (Merck, N3020). After 1 min wash in running tap water again, tissue sections were dehydrated in ethanol, dipped in xylene and finally coverslipped using DPX Mountant (Merck, 06522).
Measuring the thickness of the mucus layer in the colon
To preserve the mucus layer of the colonic epithelium, contact with any aqueous solution was avoided after the excision of the intestinal tissue. Without removing the faecal matter, several segments of the colon were cut using a scalpel and fixed overnight at room temperature in methacran/Carnoy’s solution which was composed of 60% methanol, 30% chloroform, and 10% glacial acetic acid. On the second day, fixed tissues were processed in 100% methanol (2 times, 30 min each), 100% ethanol (3 times, 60 min each) and xylene (2 times, 60 min each). Processed tissues were embedded in paraffin and 4 µm thick sections cut and stained with Alcian blue as described above. Stained tissues were photomicrographed at 60× magnification on an Olympus BX51 brightfield microscope. For both NMRs and mice, 30 independent measurements of the mucus layer were taken from 3 animals using the ‘measure’ tool in Fiji package88.
Alkaline phosphatase staining
Tissue sections on SuperFrost Plus slides (VWR, 6310108) were deparaffinized in xylene (2 times, 5 min each) and rehydrated in 100%, 90%, 70% ethanol (5 min each) and distilled water (5 min). A hydrophobic barrier was drawn around the tissue sections using a PAP pen (Vector Lab, H-4000) before incubating in the AB solution (AP Staining kit, SystemBio, AP100B-1) for 20 min at room temperature in the dark. All sections were then washed in 1× PBS (5 min, on a shaker), counterstained with Nuclear Fast Red (5 min), washed in running tap water (1 min), dehydrated in ethanol, dipped in xylene and finally coverslipped with DPX Mountant (Merck, 06522).
Immunohistochemistry
4 µm thick formalin-fixed paraffin-embedded (FFPE) sections were cut using a microtome and dried overnight on SuperFrost Plus slides (VWR, 6310108). Tissue sections were baked at 60 °C for 1 h the next day, deparaffinized in 3 rounds of xylene (5 min each) and rehydrated in 100%, 90%, 70% ethanol and distilled H2O (5 min each). Endogenous peroxidase activity was quenched by incubating sections in 3% H2O2 (Merck, 8222871000) for 20 min. A heat mediated antigen retrieval was performed by boiling sections in 10 mM sodium citrate buffer (pH 6.0) for 10 min which was followed by 20 min of cooling down in the same solution. This was followed by incubating the tissue sections in 1× PBSTX (0.1% Triton X) for 10 min. All sections were then blocked for 1 h at room temperature using 5% serum which matched the species of the secondary antibody. Next, primary antibodies were diluted in antibody diluent (1% BSA dissolved in 1× PBS) which was applied to the tissue sections and incubated overnight at 4 °C. The primary antibodies used in this study were Chromogranin A (Abcam, ab15160) at 1:2000 and BrdU (Abcam, ab6326) at 1:500. It is noteworthy that in our BrdU staining, we did not use HCl-mediated DNA denaturation and only performed heat-mediated antigen retrieval (98-100 °C) which has been shown to produce a brighter signal than acid hydrolysis89. After 3 rounds of washes (5 min each) with 1× PBST (0.1% Tween20 in 1× PBS), tissue sections were then incubated for 1 h at room temperature with biotinylated secondary antibodies diluted at 1:300. For our study specifically, we used goat anti-rabbit IgG (Vector Laboratories, BA-1000) and goat anti-rat IgG (Abcam, ab207997). To detect the biotinylated target, we used the Avidin/Biotinylated enzyme Complex (ABC) kit (Vector Laboratories, PK-6101) and developed the signal using the DAB (3,3’-diaminobenzidine) solution (R&D systems, 4800-30-07). The tissue sections were then counterstained with Harris Haematoxylin (Merck, HHS32) for 5 s, dehydrated in 70%, 90% and 100% ethanol for 15 s each, dipped in xylene and coverslipped using DPX Mountant (Merck, 06522).
mRNA ISH
Species-specific RNAscope probes from ACD Bio-techne were used to detect Lgr5 mRNA expression in NMR (584631), mouse (312171) and human (311021) intestinal tissues. We used the RNAscope Multiplex Fluorescent Detection Kit v2 (ACD Bio-techne, 323110) and followed the instructions of the manufacturer (document number 323100-USM, ACD Bio-techne) to detect Lgr5 mRNA targets at a single cell level in FFPE tissue sections mounted on SuperFrost Plus slides (VWR, 6310108).
Multiplex mRNA ISH with immunofluorescence
To enable multiplexing of mRNA and proteins, we adapted the manufacturer’s instructions (document number 323100-USM, ACD Bio-techne) for RNAscope Multiplex Fluorescent Detection Kit v2 (ACD Bio-techne, 323110) to exclude the step involving protease treatment. Once the mRNA signal was developed, we proceeded to detect proteins by first washing tissue sections (2 times, 2 min each) in 1× TBST (0.1% Tween20 in 1× Tris-buffered saline). This was followed by blocking for 1 h at room temperature with 10% serum which matched the species of the secondary antibodies. Multiple primary antibodies (diluted in 1% BSA in 1× TBS) were then applied to the tissue sections and incubated overnight at 4 °C. The dilutions of various primary antibodies used in our study were 1:500 for EpCAM (Abcam, ab71916), 1:500 for Ki67 (Cell Signaling, 12202), 1:200 for p27Kip1 (Cell Signaling, 3686 and 2552), 1:500 for BrdU (Abcam, ab6326) and 1:2000 for PHH3-S28 (Abcam, ab32388). Following primary antibody incubation, the next day we washed the sections thrice in 1× TBST (5 min each) before incubating them with fluorophore-linked secondary antibodies (at 1:500 dilution) for 1 h at room temperature. Fluorescent secondary antibodies used in our study included goat anti-rabbit Alexa 488 (Invitrogen, A11008), goat anti-rat Alexa 488 (Invitrogen, A11006), goat anti-rabbit Alexa 555 (Invitrogen, A21428) and goat anti-rabbit Alexa 633 (Invitrogen, A21070). Following the secondary antibody incubation, tissue sections were washed three times in 1× TBST (5 min each) and counterstained with DAPI (Invitrogen, D1306) for 15 min at room temperature before mounting with coverslips (VWR, 631-0138) using Diamond Antifade Mountant (Invitrogen, P36961).
TUNEL assay
Click-iT™ Plus TUNEL Assay Kit (Invitrogen, C10617) was used following the manufacturer’s instructions to detect apoptotic cells FFPE tissue sections.
EdU detection
EdU-Click 488 kit (Base Click, BCK-EdU488-1) was used according to the instructions provided by the manufacturer to detect EdU-positive cells in FFPE tissue sections.
Measuring plasma BrdU concentration
Plasma BrdU concentration was determined following the protocol described by Barker et al.90. In brief, 100 µL naked mole rat blood was collected by a tail vein puncture after 8 hand 16 h of BrdU injection. The blood was mixed with heparin to stop clotting and centrifuged at 13,000g for 15 min to separate all blood cells. Plasma was collected from the top layer and stored at −80 °C.
HEK293T cells (ATCC, CRL-3216) were cultured in high-glucose DMEM (Merck, D6546) containing 10% FBS (Gibco, 10270), 1× Penicillin-Streptomycin (Merck, P4333-100ML), and 2 mM l-glutamine (Gibco, 25030-024) at 37 °C with 5% CO2. Cells were plated on a 13 mm sterile glass coverslip precoated with poly l-lysine (VWR, 631-0149) in a 24-well plate (Starlab, CC7682-7524) and cultured overnight. The media was replaced with 500 µL fresh culture media containing 10 µL plasma or standard BrdU solution (3, 10, 20, 30, 40, 50 µg/ml) and incubated at 37 °C for 4 h. Cells were then washed with 1× PBS and fixed in 4% paraformaldehyde for 20 min at room temperature. Fixed cells were kept in 1× PBS at 4 °C before proceeding to immunocytochemical detection of BrdU.
Fixed cells on coverslips in 24 well plates were incubated with 3% H2O2 for 10 min at room temperature. After washing with 1× PBS, cells were incubated in 2 N HCl for 1 h at room temperature to denature DNA strands. Fixed cells were then incubated in 0.1 M Borate buffer (pH 8.5) for 30 min at room temperature and in 1× PBSTX (0.1% Triton X) for 10 min. Cells were blocked with 5% goat serum for 1 h at room temperature and incubated with rat anti-BrdU primary antibody (Abcam, ab6326, 1:2000) overnight at 4 °C. The next day, cells were washed three times in 1× PBST and incubated with goat anti-rat-biotin-linked secondary antibody (Abcam, ab207997, 1:400) for 1 h at room temperature. The biotinylated signal was developed using the ABC Kit (Vectastain, PK-6101) following the manufacturer’s instructions and detected with DAB solution (R&D systems, 4800-30-07). Gill’s No. 3 Haematoxylin (Merck, GHS316-500ML) was used for counterstaining and cells on the coverslips were mounted on glass slides using Aquatex mounting agent (Merck, 108562).
Crypt-villous isolation and qRT-PCR
Intestinal tissue was washed with PBS, cut open longitudinally and laid flat on a glass slide with the luminal side facing upward. The small intestinal villi were scrapped off the flat tissue by a glass slide and collected in cold 1× PBS. The remaining tissue containing crypts was chopped into <2 mm pieces using a scalpel, washed three times with ice-cold 1× PBS and incubated in chelation medium (2 mM EDTA in 1× PBS without Ca2+ and Mg2+, Gibco 10010023) for 40 min with agitation at 4 °C. The digested tissue was shaken vigorously for 30 s in 1× PBS to release crypts and villi. To separate out crypts and villi of the small intestine, the solution was passed through a 100 µm cell strainer. The isolated crypts in the flow through were pelleted and transferred to RLT Buffer (Qiagen, 79216). RNeasy microkit (Qiagen, 74004) was used for RNA extraction. Extracted RNAs were incubated with DNase1 (ThermoFisher, EN0521) at 37 °C for 30 min, followed by a 10 min incubation with EDTA at 65 °C. High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814) was used to generate complementary DNA from total RNA. Quantitative real-time-PCR (qRT-PCR) was performed on LightCycler96 (Roche) with mouse and naked mole rat Gapdh used as an endogenous control. The IDs of Taqman Gene expression assays (Applied Biosystems) used in this study are Gapdh (Mm99999915_g1, Hg05064520_gH), Muc2 (Mm01276681_m1, Hg05250665_g1), Synaptophysin (Mm00436850_m1, Hg05249763_m1), and Aldolase B (Mm00523293_m1, Hg05103981_m1). The 2-ΔΔCt method was used to calculate the relative gene expression levels.
Brightfield microscopy
Brightfield images of tissue sections were captured using an Olympus BX51 microscope coupled with an Olympus DP70 camera system using DP controller software. Villi were imaged using 10× objective while crypts were imaged with 20× (for colon) or 60× (for small intestine) objective lens. Histopathological scoring in this study was performed based on the digital images obtained on Hamamatsu (Nanozoomer HT) scanner at 40× magnification.
Histological quantification (brightfield)
To quantify cell numbers in crypt-villous structures from brightfield images, ‘cell counter’ plugin of Fiji software was used. The dimensions of crypt-villous structure were calculated using the ‘measure’ tool in Fiji.
Fluorescent microscopy
Fluorescent images of intestinal crypts were acquired from 4 µm thick tissue sections with a Plan Apochromat 63× or 100× 1.4 oil objective on a Zeiss LSM 780 upright or inverted confocal microscope. Images were acquired in Zen SP7 FP3 (black) software using 405 nm, 488 nm, 561 nm, and 633 nm laser lines in sequential tracks. Z-stacks of 6-12 optical sections with 50% overlap between subsequent planes were captured within the span of a single cell at 0.3 µm z-distance, 0.087 µm pixel dimension, and 12-bit depth.
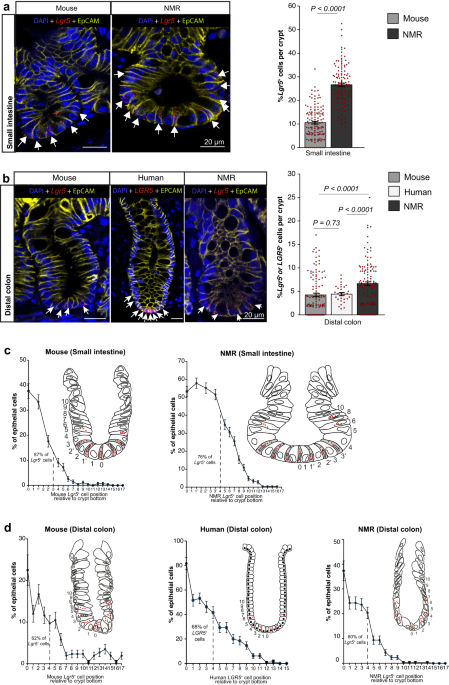
For generating the RGB images used in the figures (Figs. 1a, b, 2a–d, 3d, 4d, 7a, d, Supplementary Figs. 1–4, 5d, e, 11b), the original.czi raw files were imported into Fiji software package and a maximum intensity z-projection was created from the stacks. Using the “split channel” option of Fiji, the multicolour fluorescent images were separated into individual channels (DAPI, Alexa 488, Cy3, Alexa 633). The maximum and minimum displayed pixel values of individual channels were adjusted across the entire image set including in negative controls (i.e. linear adjustment) to correct for autofluorescence that had been introduced in the image stacks during acquisition. Then, using “merge channel” option in Fiji, two/more channels were combined to create a composite image (Lgr5/Ki67 or LGR5/KI67, Lgr5/EpCAM or LGR5/EPCAM, Lgr5/p27 or LGR5/P27, Lgr5/BrdU, Lgr5/pHH3 or LGR5/PHH3) while keeping the individual channels intact. Finally, all the individual and composite images were converted into ‘RGB color type’ and saved in TIFF format. These images (TIFF) were compiled in Adobe Illustrator 2020 software to produce the panels presented in the figures.
Histological quantification (fluorescent)
Z-stack images were processed in batch mode of Fiji package. Firstly, a maximum intensity projection was created to generate a 2D image from the stacks. Next, each channel of the image was separated, and maximum and minimum displayed pixel values were adjusted across the entire image set including negative controls. To quantify the number of rodent Lgr5 or human LGR5 mRNA expressed in a single cell, all the ISH dots were manually counted within the cell periphery demarcated by EpCAM staining. As the Lgr5 or LGR5 signal was captured using confocal microscopy at a resolution of 237 nm, overlapping/merged Lgr5 or LGR5 mRNA signal dots were rarely observed. To calculate the distribution of Lgr5+ or LGR5+ cells relative to other cells along the crypt axis, the cell present at the crypt apex was assigned position 0 and we counted cells on each side of this cell to acquire datapoints in our quantifications. Any cell containing more than three Lgr5 or LGR5 mRNA puncta was considered positive for Lgr5 or LGR5 expression (Lgr5+ or LGR5+).
We observed significant variation in autofluorescence levels between mouse, human and NMR intestinal tissues, with mouse tissue emitting the most and naked mole rats the least. This variation necessitated adjusting the laser powers of the confocal microscope during image acquisition so that maximal image contrast was achieved while also reducing the autofluorescence signals. The maximum and minimum displayed pixel values of individual channels were adjusted across the entire image set (i.e. linear adjustment), including in negative controls, to correct for autofluorescence. These adjustments resulted in varying intensities for specific signals in the three species and, therefore, we took a binary approach for the quantification of the antibody-based signals. The presence of any specific signal in the target compartment inside a cell was considered positive regardless of the staining intensity.
Estimating the length of the total cell cycle (TT) and S-phase (Ts) by cumulative labelling with BrdU
We determined the length of the cell cycle (TT) and S-phase (TS) in CBC cells (Lgr5+CBC) of naked mole rats by counting the fraction of BrdU-labelled Lgr5+CBC cells after successive pulsing over 5 days in NMRs and 2.25 days in mice. As the CBC cells (Lgr5+CBC) cells are on average asynchronously and asymmetrically dividing45, the labelling index (LI) which provides the ratio of labelled cells to the total population (LI = Lgr5+CBCBrdU+/Lgr5+CBC) at any given time (t) can be modelled by Eq. 1 below where TT is the total cell division time33.
$${{{{{rm{LI}}}}}}= (1/{{{{{rm{T}}}}}}_{{{{{rm{T}}}}}}){{{{{rm{X}}}}}}t+({{{{{rm{T}}}}}}_{{{{{rm{S}}}}}}/{{{{{rm{T}}}}}}_{{{{{rm{T}}}}}}),{{{{{rm{for}}}}}},{t}{{{{{rm{le }}}}}}{{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}-{{{{{{rm{T}}}}}}}_{{{{{{rm{S}}}}}}} {{{{{rm{LI}}}}}}= 1,{{{{{rm{for}}}}}},t > {{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}-{{{{{{rm{T}}}}}}}_{{{{{{rm{S}}}}}}}$$
(1)
Equation 1 assumes that there are no or only very few stem cells (based on p27 negativity in NMR and mouse Lgr5+CBC cells) that remain quiescent for the duration of the BrdU experiment. The lfit tool in STATA was used to calculate the least square fit of the data by considering the time points before LI reached saturation. We derived TT from the slope of the regression (TT = 1/slope). When t = 0, LI0 = TS/TT which is the y-intercept of the graph. Thus, the duration of S-phase (TS) was estimated from the y-intercept of the regression line.
Estimating the duration of the specific cell cycle phases
For human LGR5+CBC cells, we assumed KI67 is undetectable at G1/S transition and detected in the S to M phases of the cell cycle46. We determined the fraction of LGR5+CBC cells that expressed KI67 and calculated the length of S, G2 and M-phase (T(S, G2, M)) using Eq. 2:
$${{{{{{rm{T}}}}}}_{{({{{{{rm{S}}}}}},{{{{{rm{G}}}}}}2,{{{{{rm{M}}}}}})}}}{{{{{rm{KI}}}}}}67^{+}={{{{{{{rm{T}}}}}}}_{{{{{rm{T}}}}}}^{({{{{{{rm{Ref}}}}}}},31)}}{{{{{rm{X}}}}}},{{{{{{rm{LGR}}}}}}5}^{+{{{{{rm{CBC}}}}}}}{{{{{rm{KI}}}}}}67^{+}/{{LGR}5}^{+{{{{{rm{CBC}}}}}}}$$
(2)
The time in mitosis (TM) was calculated after quantifying the fraction of rodent (mouse or NMR) Lgr5+CBC or human LGR5+CBC cells positive for phospho-histone H3 using Eq. 3:
$${{{{{{rm{T}}}}}}{{{{{rm{M}}}}}}}^{{{{{{rm{Ki}}}}}}67+}={{{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}}^{({{{{{rm{linear}}}}}},{{{{{rm{regression}}}}}})}{{{{{rm{X}}}}}},{{Lgr}5}^{+{{{{{rm{CBC}}}}}}}{{{{{rm{pHH}}}}}}3+({{{{{rm{Ser}}}}}}28)/{{Lgr}5}^{+{{{{{rm{CBC}}}}}}}$$
(3)
or
$${{{{{{{rm{T}}}}}}}_{{{{{{rm{M}}}}}}}}^{{{{{{rm{KI}}}}}}67+}={{{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}}{({{{{{rm{ref}}}}}}31)}{{{{{rm{X}}}}}},{{LGR}5}^{+{{{{{rm{CBC}}}}}}}{{{{{{rm{PHH}}}}}}3}^{+}({{{{{rm{Ser}}}}}}28)/{{LGR}5}^{+{{{{{rm{CBC}}}}}}}$$
Using TS estimated by Ishikawa et al.31 previously, the length of G2-phase (TG2) was calculated using Eq. 4:
$${{{{{{{rm{T}}}}}}}_{{{{{{rm{G}}}}}}2}}^{{{{{{rm{KI}}}}}}67+}={{{{{{rm{T}}}}}}}_{({{{{{rm{S}}}}}},{{{{{rm{G}}}}}}2,{{{{{rm{M}}}}}})}{{{{{{rm{KI}}}}}}67}^{+}-left({{{{{{{rm{T}}}}}}}_{{{{{{rm{S}}}}}}}}^{{{{{{rm{KI}}}}}}67+}+{{{{{{{rm{T}}}}}}}_{{{{{{rm{M}}}}}}}}^{{{{{{rm{KI}}}}}}67+}right)$$
(4)
After quantifying the fraction of LGR5+CBC cells expressing P27, we calculated the time spent in G0 and G1 (T(G1, G0)P27+) using Eq. 5:
$${{{{{{{rm{T}}}}}}}_{({{{{{rm{G}}}}}}1,{{{{{rm{G}}}}}}0)}}^{{{{{{rm{P}}}}}}27+}={{{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}}{({{{{{rm{ref}}}}}}31)}{{{{{rm{X}}}}}},{{LGR}5}^{+{{{{{rm{CBC}}}}}}}{{{{{rm{P}}}}}}27+/{{LGR}5}^{+{{{{{rm{CBC}}}}}}}$$
(5)
We took the fraction of LGR5+P27+ cells in G0 phase (QF) from Ishikawa et al. 31 to calculate the length of G0 in human LGR5+CBC cells using Eq. 6:
$${{{{{{{rm{T}}}}}}}_{{{{{{rm{G}}}}}}0}}^{{{{{{rm{P}}}}}}27+}={{{{{{rm{QF}}}}}}}{({{{{{rm{ref}}}}}}31)}{{{{{rm{X}}}}}},{{{{{{{rm{T}}}}}}}_{(G1,G0)}}^{{{{{{rm{P}}}}}}27+}$$
(6)
Finally, using Eq. 7, we quantified the time human colonic LGR5+CBC cells spend in G1 (TG1):
$${{{{{{rm{T}}}}}}}_{{{{{{rm{T}}}}}}}={{{{{{{rm{T}}}}}}}_{{{{{{rm{G}}}}}}0}}^{{{{{{rm{P}}}}}}27+}+{{{{{{{rm{T}}}}}}}_{{{{{{rm{G}}}}}}1}}^{{{{{{rm{P}}}}}}27+}+{{{{{{{rm{T}}}}}}}_{{{{{{rm{S}}}}}}}}^{{{{{{rm{KI}}}}}}67+}+{{{{{{{rm{T}}}}}}}_{{{{{{rm{G}}}}}}2}}^{{{{{{rm{KI}}}}}}67+}+{{{{{{{rm{T}}}}}}}_{{{{{{rm{M}}}}}}}}^{{{{{{rm{KI}}}}}}67+}$$
(7)
In NMR and mouse, Lgr5+CBC cells are negative for p27 such that TG0 = 0. For these species, we derived the combined length of time spent in G1 and G2 (TG1+TG2) from Eq. 7.
Estimating cell division time of Lgr5
+above crypt base cells from single time point analysis of BrdU labelling
Using the length of TS from cumulative BrdU labelling in Lgr5+CBC cells and assuming no change in TS in Lgr5+ cells located at different positions within the crypt31, we measured the total cell division time (TT) of Lgr5+above crypt base cells using Eq. 1 by measuring the labelling index (LI) at a single time point (t) after pulsing animals with BrdU in vivo. More specifically, in C57BL/6 mice (n = 3 animals, 4 months old), we administered BrdU once and analysed intestinal tissue at t = 0.5 h. In NMRs (n = 3 animals, 6-24 months-old), we pulsed the animals with BrdU every 8 h and analysed the intestine after t = 1 day.
Statistical analysis
We used Microsoft Excel (v16.77.1) for inputting raw data after collection. All statistical tests and graphs displayed in this paper were generated using StataMP 14.1. Details of statistical tests performed are described in figure legends. P-values are generated by conducting two-tailed t-tests, F-test and Wilcoxon rank sum test as indicated in each figure legend. No blinding and randomization were performed during the analysis.
Illustration
All the figures presented in this manuscript were prepared using Adobe Illustrator 2020 (version 24.1). Vector line arts shown in Figs. 1c, d, 3a, h, 4a, h, 6a, Supplementary Figs. 5d, e, and 9a, b were created using the curvature tool of Adobe Illustrator.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s41467-023-44138-6