sgRNA design and selection
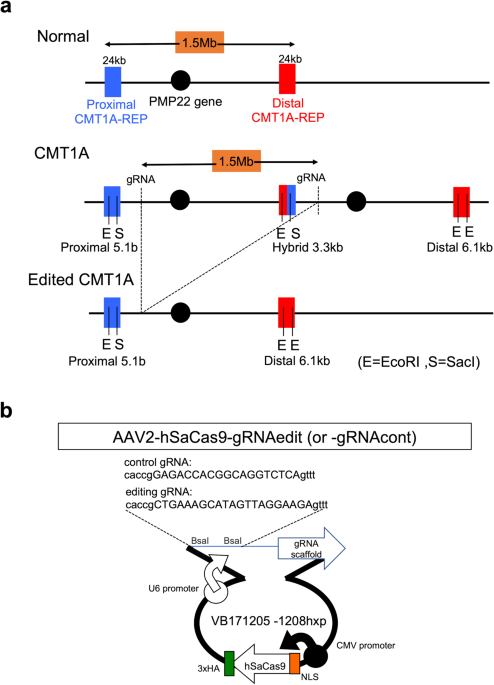
To target the genomic region of PMP22, a series of sgRNAs were designed by inserting the region between the STR polymorphic genetic markers D17S261 and D17S122 (sgRNA region: 15360587 to 15398063)26 into Cas-OFFinder (www.rgenome.net/cas-offinder). This region is outside of proximal CMT1A-REP (15575936 to 15587412), distal CMT1A-REP (14179212 to 14190596), and the PMP22 gene (15229777 to 15265357). The following five sequences were selected and subcloned into the pX601 vector (Addgene plasmid #61591) at the BsaI site after addition of a specific protospacer adjacent motif (PAM) sequence for SaCas9:
CMT1AsgRNA1-f: CACCGATATCACTCTCATGACTAGTT
CMT1AsgRNA2-f: CACCGGGCCCAAGGTCTAATTTACAT
CMT1AsgRNA3-f: CACCGCTGAAAGCATAGTTAGGAAGA
CMT1AsgRNA4-f: CACCGAACAGGGAAACAAACAGTGGG
CMT1AsgRNA5-f: CACCGAGAACTGAAAGCATAGTTAGG
The plasmid sequences were validated by the sequencing primer: pX601-sequencing: GAGGTACCTGAGGGCCTATT
For final selection from the five gRNA candidates, a T7E1 mismatch detection assay27 was used to determine the genome editing efficiency of CRISPR-SaCas9 as indicated by the InDel mutation rate. To calculate the percent cutting efficiency of the CRISPR locus, the intensity of the PCR amplicon band and digested bands were measured with Image J (http://imagej.nih.gov/ij/; National Institutes of Health, USA), and %InDel was calculated by the following formula28:
$$% {{{{{rm{InDel}}}}}}=left(1-sqrt{1-frac{left(a+bright)}{left(a+b+cright)}}right)times 100$$
(1)
AAV construction
AAV2 is considered one of the most efficient serotypes for transduction of human Schwann cells29, so was used it in this study. An AAV Helper-Free System (Agilent Technologies, Santa Clara, California, United States, Catalog #240071) was used to produce AAV particles. The recombinant AAV vectors were produced by transient transfection of HEK293 cells using a vector plasmid; a plasmid for AAV2 Rep and AAV2 Cap expression; and an adenoviral helper plasmid, pHelper. To verify AAV infection efficiency in Schwann cells, the S16 rat Schwann cell line was seeded in an 8-well glass chamber at 4 × 103 cells/well. Cells were infected with AAV1 virus containing the CAG promoter and EGF (AAV1-CAG-EGF, 2.2 × 1011 vg/mL), at MOIs of 5000, 10,000, 20,000, 50,000, 100,000, or 200,000 and incubated for 24, 48, 72, 96, 120, and 144 h.
iPSC Schwann cell differentiation
201B7 iPSCs and HPS0426 PMP22 duplication iPSCs, whose mycoplasma infection was denied by DNA staining (Hoechst 33258) and nested-PCR methods, were differentiated by modifying the previous method30. Briefly, six-well plates were coated with Matrigel GFR diluted 1:20 in DMEM/F12 at RT for 1 h. On day 0, iPSCs were seeded at 2 × 105 cells/well in Stemfit medium containing Y-27632, a ROCK inhibitor. On the subsequent day, medium was replaced by neural differentiation medium containing 1:1 DMEM/F12: Neurobasal media containing 1:100 N2, 1:50 B27, 0.005% BSA, 2 mM GlutaMAX, 0.11 mM β-Mercaptoethanol, 3 µM CHIR, and 20 µM SB. After 6 days, the cells reached 80–90% confluency and were ready for the first passage into Schwann cell precursor differentiation medium containing equal amounts of DMEM/F12 medium (#11320033 Gibco) and Neurobasal medium (#21103049 Thermo Fisher Scientific) supplement for growth and expression of neuroblastomas (N2, #17502-048 Thermo Fisher Scientific), neuronal cell culture supplement (B27 #17504044 Thermo Fisher Scientific), 0.005% BSA (Bovine Serum Albumin), 2 mM GlutaMAX (#35050061 Thermo Fisher Scientific), 0.11 mM β-Mercaptoethanol (#21985023 Gibco), 3 µM GSK3 inhibitor (CHIR99021, #252917-06-9 Tocris Bioscience), 20 µM TGF-βinhibitor (SB431542, #13031 Cayman), and 50 ng/mL Neuregulin 1 (NRG1, #396-HB-050 R&D Systems). After 18 days, the medium was changed to Schwann cell differentiation medium containing low-glucose DMEM, 1% FBS, 200 ng/mL NRG1, 4 µM Forskolin (#F6886-10MG Sigma), 100 nM RA, and 10 ng/mL PDGF-BB. Media was changed daily for 4 days.
Cells were subsequently incubated in Schwann cell differentiation medium without FK or RA for 2 days. PDGF-BB was then removed, and cells were incubated in low-glucose DMEM (1 mg/mL), 1% FBS, and 200 ng/mL NRG1 (Neuregulin #396-HB-050 R&D Systems). Immunohistochemistry was performed to verify Schwann cells marker expression in differentiated cells.
Cells were infected with AAVs one day after starting iPSC culture (day 1), one day after initiating differentiation into Schwann cell precursors (day 26), and on day 43, after differentiation into Schwann cells. Each AAV was infected at MOI:10,000. DNA from the infected cells was collected on day 50, and the genome-editing efficiency was measured using qPCR.
CMT1A editing
AAV infection of CMT1A-iPSCs was performed as follows. Twelve-well plates were coated with Matrigel and seeded with CMT1A iPSCs (7.5 × 104 cells/well). Four wells contained cells without virus, four wells cells were infected with AAV encoding control gRNA (AAV2-hSaCas9-gRNAcont, MOI 10,000), and four wells were infected with AAV2-hSaCas9-gRNAedit, MOI 10,000. After 1 week, PCR for AAV-ITR and Nested qPCR were performed to determine the efficiencies for AAV infection genome-editing, respectively.
For AAV-ITR PCR, primers from Riken and Addgene were used. Riken: 5′-GAGTGGCCAACTCCATCACTAGGGGTTCCT-3′. Addgene: fwd ITR primer 5′-GGAACCCCTAGTGATGGAGTT/ rev ITR primer 5′-CGGCCTCAGTGAGCGA. AAV-ITR PCR was conducted with LA Taq with the following cycles: 94 oC 1 min, 35 cycles of 94 oC for 1 min, 64 oC for 30 s, and 72 oC for 3 min, and a final cycle of 72 oC for 7 min.
Nested qPCR was performed according to the Human Taqman Copy Number Assay (ThermoFisher). The probe sequences are as follows:
CMT1A Tqm: 6FAM-AAGAAGAATCGTGGGCACACCACCA-TAMRA
Primers for primary PCR of CMT1A recombination site are:
CMT1A PR1: TGATATTTAAAGATTTCATGTC
CMT1A DF1: GGATTCAGAGACATTAGTGTTCC
Products from the first PCR were digested with ExoI (NEB).
Primers designed for secondary PCR of the CMT1A recombination site are below:
CMT1A PR2: CATGTCATTAGACCAAAGAaC
CMT1A DF2: AGAAACATACTAGTTGATATCTTCTaT
RNase P (VIC-TAMRA) was used as an internal control. Both PCR assays were performed with Platinum Taq (TAKARA).
Signal intensity of bands on agar gel was measured using Image J software (http://imagej.nih.gov/ij/. National Institutes of Health, USA).
Quantification of the hybrid region was conducted according to the following formula:
$${Hybrid}{{{{_}}}}{Relative}{{{{_}}}}{Quantification}=frac{{{2}^{{-({hybrid}-{internal}{{{{_}}}}{control})}_{{AAV}{{{{_}}}}{treated}}}}}{{2}^{{-({hybrid}-{internal}{{{{_}}}}{control})}_{{untreated}}}}$$
(2)
Southern blot analysis
For probe labeling, genomic DNA prepared from normal iPSCs was used for PCR (KOD Fx Neo) with the following primers: CMT1A_Probe-Fw1: AAGAAGAATCGTGGGCACAC and CMT1A_Probe-Rv1: AGTGCAAACCATGATCACCC. The PCR products were purified with the Favorgen kit and labeled with DIG-DNA labeling (Roche DIG labeling). Southern blot was performed using 5 µg iPSC genomic DNA digested with EcoRI and SacI, electrophoresed in 0.8% agar gel, treated with 0.25 N HCl for depurination and cleaved by alkaline treatment. DNA was transferred from agarose gel to a membrane (Hybond-N, RPN303 N, GE Healthcare, Chicago, IL, USA) at room temperature for 20 h. Prehybridization of the blotted membrane was performed using DIG Easy Hyb, and hybridization was performed by adding DIG-labeled DNA probe (35 ng/mL) at 42 oC for 20 h.
Immunocytochemistry
Cells were fixed with 4% formaldehyde for 10 min at 25 °C and permeabilized with 0.1% Triton X-100 in PBS for 5 min at 25 °C. After blocking with 10% Fetal Bovine Serum (#10270106, Gibco, MA, USA) for 30 min at 25 °C, cells were incubated with primary antibody for 16 h at 4 °C and with secondary antibodies for 1 h at 25 °C. The antibodies were as follows: rabbit anti-S100B antibody (1:500, ab52642, abcam, Cambridge, UK), goat anti-Sox10 antibody (1:100, sc-17342, Santa Cruz Biotechnology, Dallas, TX, USA), mouse anti-Myelin Basic Protein antibody (1:200, ab62631, abcam, Cambridge, UK), rabbit anti-MAP2 (1:100, sc-32791, Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-PMP22 (1:500, ab126769, abcam, Cambridge, UK), Alexa Fluor 647 anti-MAP2 antibody (1:500, ab225315, abcam, Cambridge, UK), Alexa Fluor 488-conjugated anti-rabbit IgG (1:1000, #A21206, Molecular Probes, Eugene, OR, USA), Alexa Fluor 568-conjugated anti-mouse IgG (1:1000, #A10037, Molecular Probes, Eugene, OR, USA) and Cy3-conjugated anti-mouse IgG (1:500, 705-165-003, Jackson Laboratory, Bar Harbor, ME, USA). Images were taken by confocal microscopy (Olympus FV1200IX83, Tokyo, Japan).
Western blot analysis
Cells were scraped and collected with PBS. After centrifugation (1500 rpm, 5 min), cell pellets were lysed in sample buffer (25 mM Tris-HCl ph6.5, 5% glycerol, 1% SDS, 1% mercaptoethanol and 0.05% BPB) and heated at 100 °C for 5 min. Samples were electrophoresed by SDS-PAGE, and the gels were transferred onto Immobilon-P polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA) using semi-dry transfer, and then blocked with 7.5% milk in TBST (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20). Then the membranes were incubated with mouse anti-PMP22 antibody (1:200, sc-515199, Santa Cruz Biotechnology, Dallas, TX, USA) for 3 h and mouse anti-GAPDH antibody (1:3000, MAB374, sigma-Aldrich, St. Louis, MO, USA) for 1 h at 25 °C. Then incubated with HRP-linked anti-mouse IgG (1:5000, NA931, GE Healthcare, Buckinghamshire, UK) for 1 h at 25 °C. ECL Select Western Blotting Detection Reagent (RPN2235, GE Healthcare, Chicago, IL, USA) and a luminescent image analyzer (ImageQuant LAS 500, GE Healthcare, Chicago, IL, USA) were used to detect proteins.
Electron microscopy
Co-cultures of iPSC-derived neurons and iPSC-derived Schwann cells were pre-fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2 h at 4 °C, and post-fixed with 1% osmiumtetroxide for 2 h at 4 °C. Following fixation, cells were dehydrated with a graded series of ethanol, and embedded in epon for 48 h at 60 °C, and 24 h at 120 °C. Ultrathin sections (80 nm) were cut with an ultramicrotome (US6, Leica, Wetzlar, Germany) and incubated with uranyl acetate and lead citrate. Sections for Immunoelectron microscopy were incubated in blocking buffer (1% BSA in PBS) for 1 h at 25 °C, and stained with rabbit anti-Crispr-Cas9 antibody (1:25, ab203933, abcam, Cambridge, UK) for 16 h at 4 °C, then incubated with anti-IgG(H + L), Rabbit, Goat-Poly, Gold 10 nm, EM (1:200, EMGAR10, BBI Solutions, Wales, UK), anti-IgG(H + L) for 2 h at 25 °C. Ultrathin sections were observed by electron microscopy (JEM-1400, JEOL, Tokyo, Japan).
Whole genome sequencing
For evaluation of off-target effects of genome editing vector in vitro, genome DNA was extracted from control and genome-edited iPS cells according to the protocol described above. For evaluation of off-target effects by intraneural injection of genome editing vector in vivo, 6 × 1010 vg of AAV2-hSaCas9-gRNAedit was injected to sciatic nerve of three C57/BL6 mice at 3 months of age, and the injected tissues were dissected after 4 weeks. FavorPrep Tissue Genomic DNA Extraction Mini kit (FATGK001, FAVORGEN, Ping Tung, TAIWAN) was used to extract genome DNA from iPS cells as well as AAV-injected or control non-injected mouse tissues. Whole genome sequencing (WGS) was performed by Illumina NovaSeq 6000 with Truseq DNA Nano in Rhelixa (Tokyo, Japan), at the condition of 150 bp×2 paired-end (PE150), 90 G bases per sample, and 600 M reads per sample (300 M pairs).
The acquired data were analyzed by using HaplotypeCaller function in Genome Analysis Toolkit (GATK, v4.2.3.0), and the results were annotated by using Ensembl Varient Effect Predictor (VEP) (https://asia.ensembl.org/info/docs/tools/vep/index.html)31 to identify known and unknown SNPs different from the data base (human iPS cells: dbSNP, C57/BL6 mice: Ensembl Variation). Original SNPs found in the non-infected controls were further excluded to identify candidate SNPs for de novo mutations by genome editing. Genome regions duplicated in human CMT1A iPS cells were identified from the increase of reads in WGS. To visualize the increase of reads, IGVTools (Broad Institute, v2.5.3) was used to calculate read depth from WGS data.
TIDE for evaluation of genome editing efficiency
TOPO subcloning and Sanger sequencing were performed following the protocol (https://www.thermofisher.com/jp/ja/home/life-science/genome-editing/genome-editing-learning-center/genome-editing-resource-library/genome-editing-application-notes/sanger-sequencing-facilitate-crispr-talen-mediated-genome-editing-workflows.html). Genomic DNA prepared from CMT1A-iPSCs 2 weeks following AAV2-hSaCas9-gRNAedit infection was amplified by junction 4F (TGGATGGTGGTAGGTATCATTCA) and Junction 4R (TGGGGCACATGAGATATTTTGG) primers. The amplified DNA fragments were subcloned into pCR®4-Blunt TOPO®, and 1,000 clones were sequenced by Sanger method at Eurofins Genomics (Tokyo, Japan). The sequence data were analyzed by BLAST at the supercomputer SHIROKANE (The Institute of Medical Science, The University of Tokyo).
Karyotype analysis
A G-band analysis was performed to determine the karyotype of the iPSC line. Twenty metaphase plates were analyzed.
Statistics and reproducibility
Statistical analyses for biological experiments were performed using Graphpad Prism 8. Biological data following a normal distribution are presented as the mean ± SEM, with Tukey’s HSD test for multiple group comparisons or with Welch’s t-test for two group comparisons. The distribution of observed data was depicted with box plots, with the data also plotted as dots. Box plots show the medians, quartiles, and whiskers, which represent data outside the 25th–75th percentile range. Data not following a normal distribution are examined by Wilcoxon’s rank sum test with post-hoc Bonferroni correction. To obtain each data, we performed biologically independent experiments. The number of samples was indicated in each figure and figure legends.
Ethics declarations
This study was performed in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the Japanese Government and the National Institutes of Health. Animal experiments were performed in accordance with the ARRIVE guidelines. All experiments were approved by The Committees for Gene Recombination Experiments, Ethics and Animal Experiments of the Tokyo Medical and Dental University (G2018-082C3, O2017-008, and A2021-211A). Normal and CMT1A iPSC (201B7 and PMP22 duplication iPSCs) were derived from Cell Bank of RINEK BRC (HPS0063 and HPS0426), and researches with these iPSCs were approved by The Ethics Committee of Tokyo Medical and Dental University (O2017-008). Informed consent was obtained from all participants for generation of iPSCs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s43856-023-00400-y